Team:UBonn HBRS/Description/Cloning
![]()
Cloning
Contents
General Abstract
The cloning aspect of this project is responsible for programming bacterial cells to secrete enzymes which can decouple ink particles from paper fibers.
To this end, genes coding for enzymes were synthesized and ligated into a plasmid backbone, which is designed for use in both Escherichia coli and Bacillus subtilis.
As expression of enzymes is intended for both Bacillus subtilis and E.coli, there are two separate tracks for cloning. For B. subtilis, the genes were assembled into pSBbs1AK1 along with secretory tags, and for E.coli, an inductive promoter (pLac) is placed upstream of the genes.
Genes of Interest
Within the scope of this project, genes of interest (GOIs) are those which catalyse either hydrolysis of paper fiber cellulose or ink particles.
Synthesized at GenScript, each of these genes includes a sequence coding for His-tag, for ease of protein purification.
Cellulase group
Enzymes which break down cellulose molecules into monosaccharides, therefore partially disintegrate paper fibers and set ink particles free into the supernatant.
| Gene name | Enzyme coded | Part number | Note |
| XynA | Xylanase | BBa_K2029002 | http://www.microbialcellfactories.com/content/11/1/66 |
| CelA | Cellulase | BBa_K2029004 | https://www.ncbi.nlm.nih.gov/pubmed/22101447 |
| EngB | Endoglucanase | BBa_K2029005 | http://www.ncbi.nlm.nih.gov/pmc/articles/PMC1785254/ |
| BsCel5c | Cellulase | BBa_K2029006 | http://www.ncbi.nlm.nih.gov/pubmed/21549854 |
Esterase-Laccase group
Enzymes which hydrolyse esters or oxidize phenols and aromatic compounds found in commercial inks. Their ability to attack ink particles is not well studied.
| Gene name | Enzyme coded | Part number | Note |
| EstC2 | Esterase | BBa_K2029007 | https://www.ncbi.nlm.nih.gov/pubmed/21619698 |
| LipA | Lipase | BBa_K2029008 | http://onlinelibrary.wiley.com/doi/10.1002/bit.10201/abstract |
| Bpul | Laccase | BBa_K2029009 | https://www.ncbi.nlm.nih.gov/nucleotide/388482909 |
General Strategy
The primary target chassis organism is B. subtilis, for which a secretory tag system was designed. At first E. coli was only intended to be used as the chassis for cloning, but as the project went on, E.coli was decided on as the backup system in case gene expression in B.subtilis fails.
In B. subtilis
As they express secretory proteins, GOIs require a signaling system in order to function in B.subtilis.
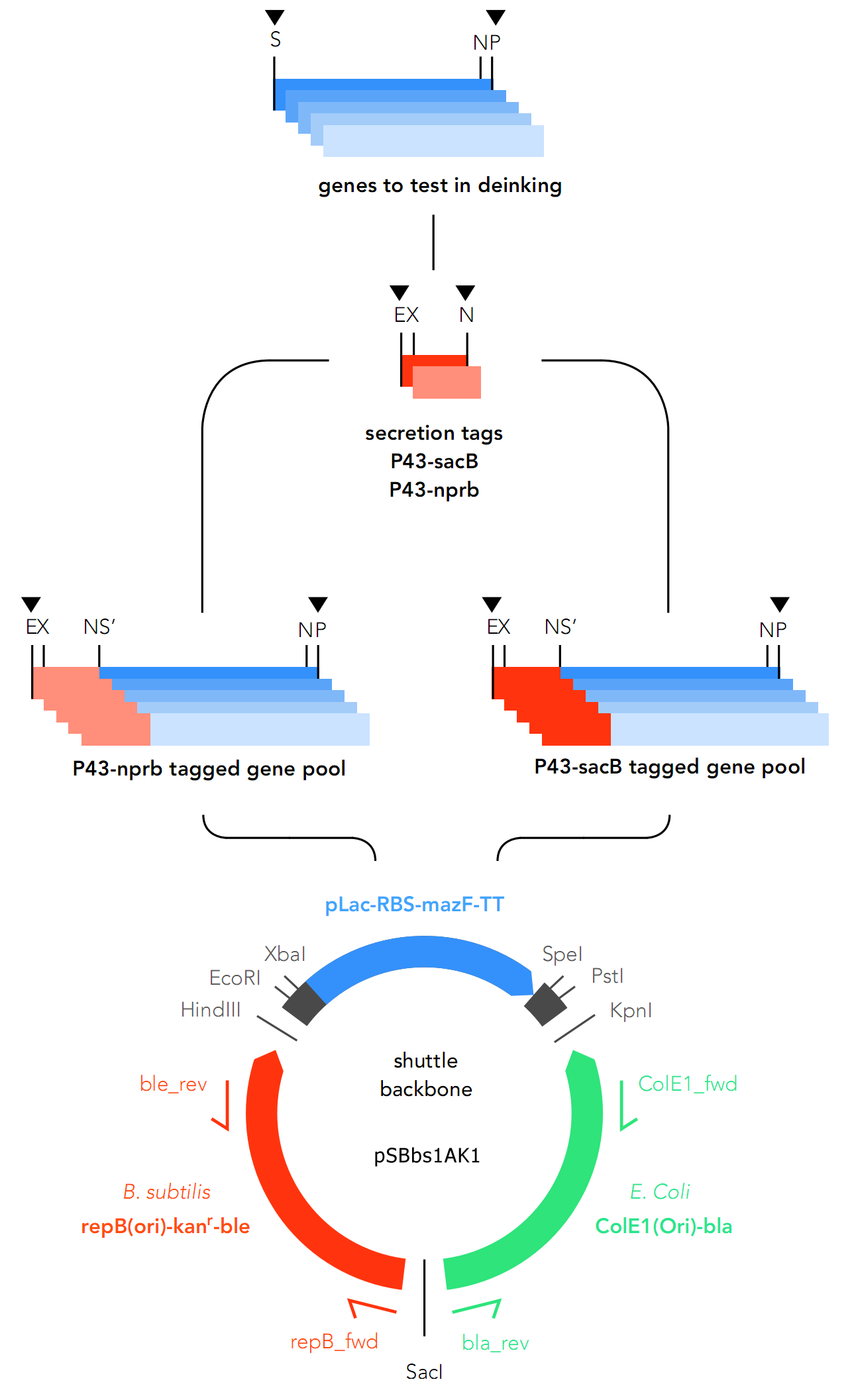
For this reason, two secretory tags are chosen to precede the genes: P34-sacB (Part: BBa_K2029001) and P34-nprb (BBa_K2029002). Both are constitutive in nature, which means they cannot be deactivated.
In E. coli
E.coli is intended to be the “Plan B” host organism, in case protein expression in B.subtilis cannot be achieved.
An inductive promoter – “pLac” – is coupled with a sequence coding for ribosome binding site (RBS). This whole sequence, so called pLac-RBS (Part: BBa_K2029014), was synthesized at Integrated DNA Technologies (IDT).
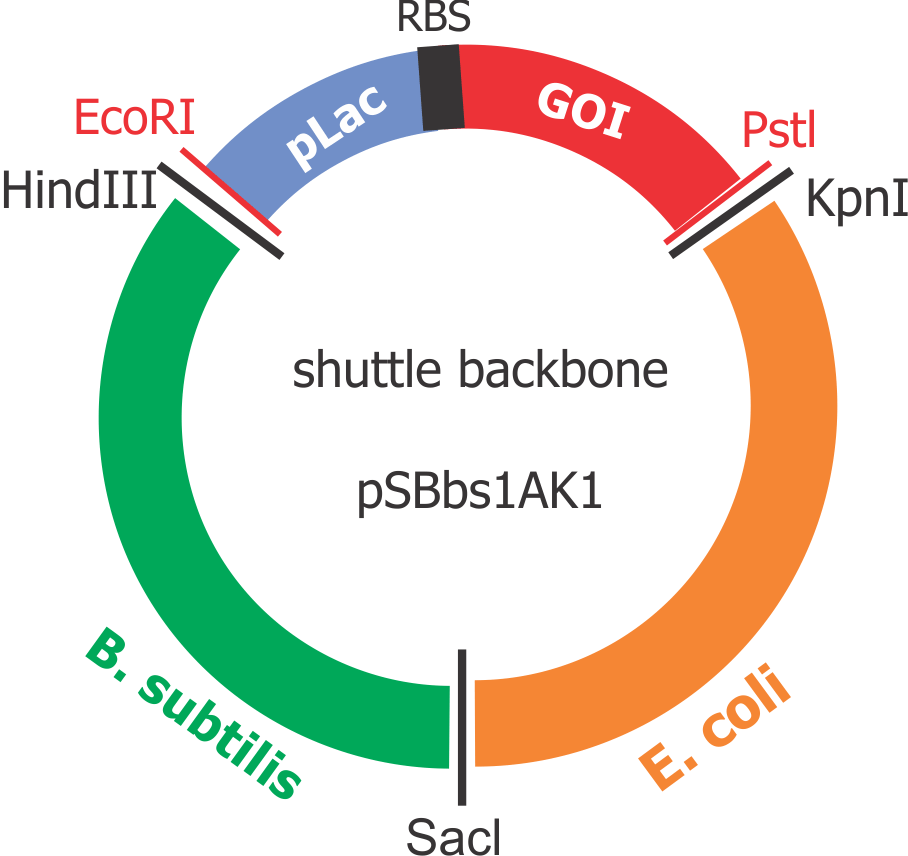
Composite “shuttle” plasmid backbone
As the GOIs are intended to be put into both chassis – E. coli and B. subtilis, a so-called “shuttle” backbone was developed to ensure compatibility with both species. This plasmid backbone - now given the name pSBbs1AK1 in the iGEM part registry – contains two different origins of replication, as well as confers resistance to two different antibiotics — one for each organism: Kanamycin resistance for B. subtilis and Ampicillin resistance for E. coli.
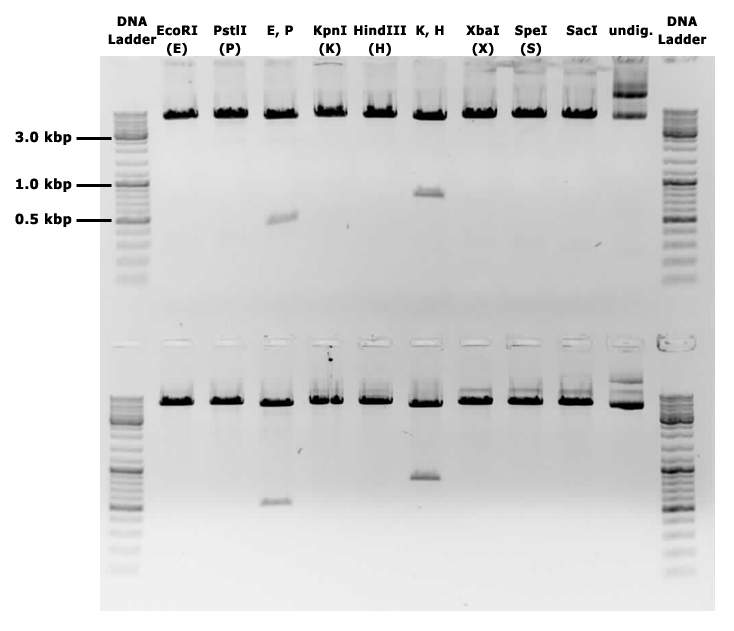
This backbone, however, has proven to be unsuitable for the gene assembly process due to its large size (appr. 5.5 kbp), hence all tag-gene constructs for B.subtilis and pLac-RBS-gene constructs for E.coli were assembled using existing iGEM backbones such as pSB1C3 and pSB4C5, and only transferred into pSBbs1AK1 in the final step.
Discussion
A major limitation of this cloning project is the lack of success, up until this point, in either aforementioned tracks for B. subtilis and E. coli.
With the B. subtilis track, while a total of 3 tag-gene constructs (Parts: BBa_K2029011, BBa_K2029012, BBa_K2029013) have been obtained, none of which has been successfully cloned into B. subtilis.
It is worth noting that all three constructs obtained contain the P34-nprb tag, and none with P34-SacB. Data showed there was little to no colony growth with pSB1C3 plasmids containing these tag-gene constructs. It is hypothesised that as these secretory tags are constitutive, protein expression overwhelms the E. coli cells used for assembly, and ultimately killed them. In a change of strategy, the tags were cloned into pSB4C5, a low-copy iGEM plasmid, which should reduce protein expression in theory. Results showed a noticeable improvement with P34-nprb, but no significant improvement in P34-SacB:
| Sample | Plate 1 | Plate 2 |
| SacB + C3 | 0 | 0 |
| SacB + C3 | 4 | 4 |
| nprb + C3 | 0 | 0 |
| nprb + C5 | 700 | 300 |
| Controls | ||
| SacB | 0 | |
| nprb | 0 | |
| C3 | 0 | |
| C5 | 0 | |
With the E. coli track, ligation of the pLac-RBS construct with GOIs did not yield any colony until the very last week of work in the laboratory, so even though colonies have appeared, it has not been ascertained whether they hold the desired plasmids.
Due to the lack of cloning success so far, no enzyme has been produced by bacterial cells, and the cloning sub-area has not been able to provide input to other aspects of the project, namely protein expression and purification in B.subtilis, deinking and enzyme assay.
Protocols used
Ligation
Masses of DNA needed are calculated to achieve as close as possible to an ideal ratio of 3:1 between molar amounts of insert and backbone. Volumes of DNA needed can then be calculated by dividing mass for concentration of sample.
Volume of 10x T4 Ligase from New England BioLabs (NEB): 10% of total mixture
Volume of Rapid 2x ligase buffer: 50% of total mixture
Total volume reaches 5 μL.
Transformation of E.Coli cells
5 μL of ligated DNA + 50 μL of chemocompetent E.coli, well mixed by resuspension with pipette
Incubation on ice for 30-45 minutes
Heat shock at 42°C for 42 seconds
Incubation on ice for 5-10 minutes
Addition of 250 μL of SOC medium
Incubation at 37°C for 30-45 minutes
Plating 100-150 μL of bacterial mixture on petri dish with correct antibiotics
Incubation at 37°C overnight
DNA purification (“prep”)
Macherey-Nagel’s NucleoSpin® Plasmid - Isolation of high- and low-copy plasmid DNA from E. coli
Gel cleanup
Macherey-Nagel’s Gel Extraction
Dephosphorylation
(to prevent re-ligation of backbones)
Thermo Scientific Fast AP Thermosensitive Alkaline Phosphatase - Protocol for nucleic acid dephosphorylation