Difference between revisions of "Team:LMU-TUM Munich/Localization"
(→Subcellular protein localization using fluorescence microscopy and western blot) |
(→An everlasting bond: creating a stable cell line) |
||
| Line 164: | Line 164: | ||
</div><!-- container --> | </div><!-- container --> | ||
| + | <div class="container-fluid"> | ||
| + | <div class="row"> | ||
| + | <div class="col-xs-12"> | ||
| + | <div class="caption"> | ||
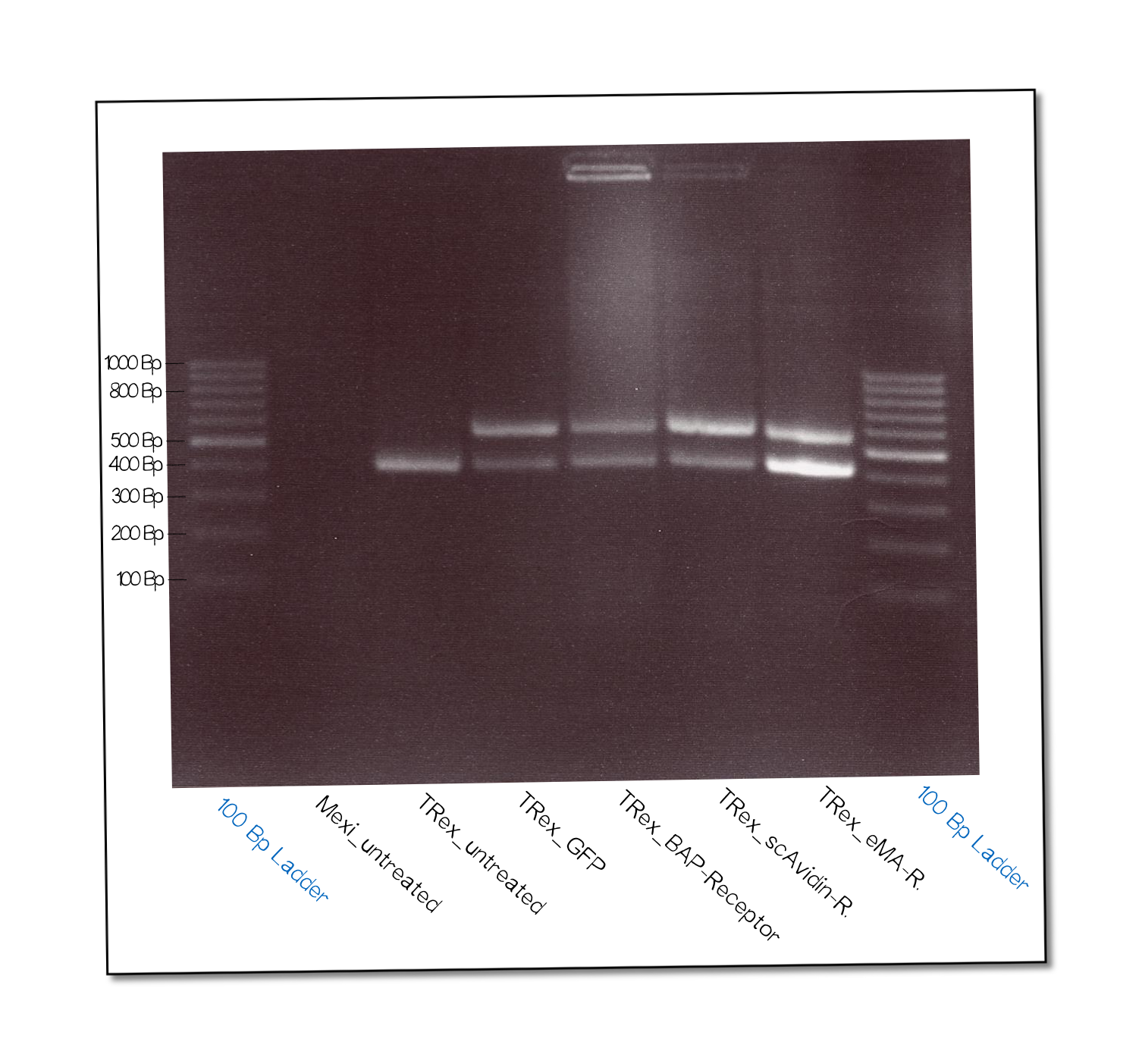
| + | The succesful integration of the pcDNA plasmids was verified by genome-PCR (see methods). Therefor we designed three primers: One that binds to the SV40 promotor and part of the following FRT site, one that binds part the hygromycin restistance gene and another one for the <i>lac</i>Z-Zeocin sequence. In the event of stable integration the size of the amplicons will be around 550 bp, according to the distance between the SV40 and the hygromycine resistance gene. Wheareas in case of no intergration the amplicon size will be around 400 since this corresponds to the distance between the SV40 and the <i>lac</i>Z-Zeocin sequences. | ||
| + | These results are shown in figure on the right. The lower band visible in the samples 2-5 leads us to the conslusion that despite hygromycine selection the cells are not yet a monoculture, something we also observed in the flow cytometry analysis further discussed below. | ||
| + | </div> | ||
| + | |||
| + | <div class="imagelink">[[Media:MUC16 GenomPCR.png]][[Image:MUC16 MUC16 GenomPCR.png|400px|rechts|link=]] | ||
| + | </div><!-- imagelink --> | ||
| + | </div><!-- col-xs-12 --> | ||
| + | </div><!-- row --> | ||
| + | </div><!-- container --> | ||
| + | |||
| + | <div class="white-box"> | ||
| + | |||
| + | The succesful integration of the pcDNA plasmids was verified by genome-PCR (see methods). Therefor we designed three primers: One that binds to the SV40 promotor and part of the following FRT site, one that binds part the hygromycin restistance gene and another one for the <i>lac</i>Z-Zeocin sequence. In the event of stable integration the size of the amplicons will be around 550 bp, according to the distance between the SV40 and the hygromycine resistance gene. Wheareas in case of no intergration the amplicon size will be around 400 since this corresponds to the distance between the SV40 and the <i>lac</i>Z-Zeocin sequences. | ||
| + | These results are shown in figure on the right. The lower band visible in the samples 2-5 leads us to the conslusion that despite hygromycine selection the cells are not yet a monoculture, something we also observed in the flow cytometry analysis further discussed below. | ||
| + | </div> | ||
*Abbildung mit stabiler Integration, Zeocin-Test, genomische PCR | *Abbildung mit stabiler Integration, Zeocin-Test, genomische PCR | ||
Revision as of 01:17, 18 October 2016

The receptors
In the following, we are going to explore the thought processes that went into the design of our receptors that polymerize cells for bioprinting.
For that purpose, we built two kinds of receptor constructs that both on their own, when being expressed in mammalian cells, are able to cross-link cells in streptavidin solution in our very own approach of bioprinting.
1) A receptor containing an extracellular biotin acceptor peptide (BAP) that is endogenously biotinylated by a coexpressed biotin ligase (BirA) and thus presents biotin groups at the cell surface.

The biotin acceptor peptide (BAP) is a 15 amino acid peptide sequence originating from E. coli. [1] Being biotinylated by the biotin ligase BirA at a lysine residue within the recognition sequence, it mediates the functionality of the receptor by presenting biotin, allowing the interaction of the cell surface with streptavidin in the reservoir solution. The biotin ligase BirA is therefore encoded by the same vector as the receptor, with an internal ribosome entry site (IRES) allowing translation of two open reading frames (ORFs) from a single polycistronic plasmid. BirA is targeted to the ER via an Igκ signal peptide as well as an ER retention signal, allowing it to biotinylate translocating proteins possessing the recognition sequence.
2) Receptors presenting an extracellular, monomeric or single-chain avidin variant.
These avidin derivates, by design, allow the functional fusion of the otherwise tetrameric avidin molecule with our receptor. Two different variants were hereby used: The so-called enhanced monomeric avidin, a single subunit avidin that is able to bind biotin as a monomer, and single-chain avidin, which resembles the naturally occuring avidin tetramer, but has the subunits being connected via polypeptide linkers.
Both receptors generally mediate the same purpose: By interacting with streptavidin in the printing reservoir - either directly by presenting biotin (biotin acceptor-peptide) or indirectly via binding to a biotinylated linker (single chain-avidin variants), cells are being cross-linked due to the polyvalent binding of single streptavidin molecules in the printing solution to several cellular biotin groups at once.
The incredible journey: signal peptides and protein targeting
Essential to a protein's function is not only its activity or binding properties, but also its presence at the right time, at the right place - in our case, the right place being the cell surface. One of the most ubiquitous elements for protein targeting is the signal peptide, which enables the translocation of proteins to the ER, from where their journey may continue to their place of function. Proteins containing a signal peptide include transmembrane proteins, secreted proteins, proteins of the ER itself, proteins of the Golgi apparatus and several more.[2] For our project, signal peptides constitute a crucial part for many constructs, being used for targeting of transmembrane proteins to the cell surface (receptors), targeting proteins to the ER (BirA) and the secretion of proteins into the surrounding medium (luciferase assays for the quantification of expression levels).

The hydrophobic signal peptide (also called start-transfer sequence) usually consists of less than 30 amino acids [4] and is recognized by the signal recognition particle upon translation at the rough ER, mediating the co-translational translocation of the nascent peptide chain into the ER.[5] During this project, three different signal peptide constructs are being tested in order to determine the one resulting in the highest expression levels:
- The EGFR signal peptide was taken from the iGEM parts registry ([http://parts.igem.org/Part:BBa_K157001:Design BBa_K157001]) and combined with a BioBrick containing the CMV promoter sequence ([http://parts.igem.org/Part:BBa_K747096 BBa_K747096]) via the RFC10 cloning standard.
- The BM40 and Igκ signal peptides were designed by us and synthesized under the aspect of adding additional spacing and functional elements to the 5'-UTR of the constructs, and combined with a BioBrick containing the CMV promoter sequence ([http://parts.igem.org/Part:BBa_K747096 BBa_K747096]) via the RFC10 cloning standard.
As shown below, the EGFR signal peptide taken from the parts registry is constructed in a way that the signal peptide ORF immediately follows the RFC10 cloning scar after the CMV promoter, thus resulting in a very short 5’ untranslated region (UTR) of the transcribed mRNA. The combination of the CMV promoter with the synthesized BM40 and Igκ signal peptides allows for a considerably longer 5’ UTR of the resulting mRNA - additionally allowing them to contain a full Kozak consensus sequence by design. The Kozak sequence is recognized by the ribosome as a translational start site; this element missing or deviating from the consensus sequence may considerably decrease translation efficiency.[6] For the EGFR signal peptide construct, a Kozak sequence is not present, as it would have to preceed the start codon ATG - a position which is occupied by the RFC10 cloning scar. Since both the distance from the promoter to the open reading frame as well as the Kozak consensus sequence are considered crucial parameters for expression levels[7], BM40 and Igκ signal peptide constructs are likely to resut in increased expression levels compared to proteins containing the EGFR signal peptide from the parts registry.
Prediction of signal peptide functionality
As the quantitative functionality of signal peptides had to be determined before including one of these into the final receptor constructs, the three options (Igκ, BM40, EGFR) were tested via bioinformatic tools as well as a secretion assay of luciferase constructs.
Bioinformatic signal peptide analysis via the SignalP server
The [http://www.cbs.dtu.dk/services/SignalP SignalP] server, being able to discriminate between the hydrophobic signal peptide sequence and the hydrophobic transmembrane domain[8], can determine whether a sequence possesses the biophysical requirements for functioning as a signal peptide. Furthermore, it is able to predict the probable cleavage site of the signal peptide after its translocation into the ER. For all constructs, signal peptide functionality is predicted, as well as a potential cleavage site. The complete translated amino acid sequences of the respective receptor constructs were used as input. The algorithm for eukaryotes with default D-cutoff value was chosen.

For all three signal peptides, the high S-score indicates high signal peptide functionality. For the Igκ signal peptide, a cleavage site between amino acid 20 and 21 within the signal peptide is predicted. For the BM40 signal peptide, a cleavage site between amino acid 17 and 18 is predicted, and for the EGFR signal peptide, a cleavage site between amino acid 24 and 25 is predicted.
Quantification of signal-peptide mediated translocation via a secretion luciferase assay
For the quantification of signal peptide functionality via a luciferase assay, three constructs were created and tested - each containing one of three signal peptides (the EGFR signal peptide, the Igκ signal peptide or the BM40 signal peptide) and a nanoluciferase as well as a CMV promoter, a Strep-tag II for immunochemical detection and the hGH polyadenylation signal sequence. Not containing a transmembrane domain, the nanoluciferase fusion protein is being translocated into the ER and then secreted into the medium. Using a luciferase assay, one can quantifiy the amount of luminescence - and thus, proportionally, the amount of secreted luciferase - by measuring the conversion of luciferin into visible light and integrating it over a timespan of 5 s. Therefore, medium samples were taken every 12 h after transfection of cells and measured via the Promega NanoGlo luciferase assay system according to the manufacturer's instructions.

Other receptor elements: Stability and detection
Apart from the functional parts that mediate the (strept)avidin-biotin interaction, other elements in the receptor were designed to make sure it is optimally translocated to the membrane, as stable as possible, and easily detectable.
EGFR transmembrane domain
For anchoring in the membrane, we decided to use a type I membrane protein, which possesses a single membrane span with defined localization of N- and C-terminus out- and inside of the cell, respectively. The N-terminal transmembrane α-helix of human EGFR (UniProt P00533, amino acids 622-653) was herefore chosen (see below the topology prediction of EGFR via the [http://www.cbs.dtu.dk/services/TMHMM/ TMHMM 2.0 server for the prediction of transmembrane helices][9]). A stop-transfer sequence consisting of charged amino acids - being characteristic for type I membrane proteins - was added at the C-terminus, and the sequence was furthermore flanked by a (GGGGC)2-linker at the N- and C-terminus, respectively. The addition of a stop-transfer sequence as in the naturally occuring EGFR sequence, as well as the addition of flexible linkers, makes our EGFR-TMD an improvement over the BioBrick [http://partsregistry.org/wiki/index.php?title=Part:BBa_K157002 BBa_K157002], which does not possess either of these elements.

Moreover, the receptor contains three functional elements for its detection: The intracellularly located red fluorescent protein mRuby 3 for detection of the receptor via fluorescence microscopy, an extracellular epitope domain for immunochemical detection via A3C5-antibody fragments and an intracellular Strep-tag II for the detection and purification via immunochemical methods.
The vector furthermore contains the poly-adenylation signal of human growth hormone (hGH) for functional polyadenylation of the transcribed mRNA. A description for the respective functional elements is given in the following sections.
A3C5 epitope tag
Antibodies have various areas of application in life science, including protein detection, pulldown experiments and even immunotherapy. Having originally been discovered in 1995 during an in vitro screening for antibodies against cytomegaloviral proteins[10], the A3C5 antibody is an established molecular tool for specific recognition of proteins. By tagging cell surface proteins with an epitope specifically recognized by A3C5 (being a peptide sequence of 11 amino acids), one is easily able to detect tagged proteins via immunofluorescence microscopy, FACS or one may purify them via pulldown experiments. Through the latter, one may also screen for in vivo interaction partners of the tagged protein.
mRuby 3
Having originally been engineered as a monomeric form of the red fluorescent protein eqFP611, mRuby variants are sine of the brightest red fluorescent proteins available. [11] With an excitation maximum at a wavelength of 558 nm and an emission wavelength maximum of 605 nm, the resulting stokes shift of 57 nm makes mRuby a good choice for fluorescent microscopy imaging and, thus, allows the visualization of the receptor in its cellular environment by being fused to the C-terminus of the transmembrane domain. mRuby is especially powerful for the fusion with receptors - not only due to its small size and high stability, but also due to the fact that, in comparison with eGFP, it appears up to 10 times brighter in membrane enviroments. The mRuby variant used here is mRuby 3, having been engineered for even more improved brightness and photostability.[12]
Strep-tag II
The Strep-tag II is an eight amino acid peptide sequence that specifically interacts with Streptavidin and can thus be used for easy one-step purification of the receptor via immunochemical methods.[13]
Nanoluciferase
The concept of bioluminescence has a great meaning for several invertebrate species, allowing communication with other individuals, signaling receptiveness or deterring predators.[14] The most common enzyme responsible for the creation of bioluminescence are the luciferases (from the latin words ’lux’ and ’ferre’, meaning ’light-carrier’). These enzymes, among others found in fireflies and deep-sea shrimp, commonly consume a substrate called luciferin as well as energy in the form of ATP or reduction equivalents in order to create light-emission through oxidation. The underlying mechanism hereby relies on the creation of an instable, highly excited intermediate under the consumption of energy. This high-energy intermediate then spontaneously falls back into a state of lower energy, emitting the energetical difference as visible light. While mainly being used as a folded, stabilizing element at the N-terminus for the construct containing the biotin acceptor peptide, the nanoluciferase could also be used for advanced binding studies via luciferase assays.[15]

Luciferases have also found their way into biotechnological applications.[16] The simplistic concept of creating visible (and thus easily measurable) light makes luciferases ideal reporters for the expression of secretable proteins via a corresponding assay. This project also made use of a luciferase for the determination of expression levels of the BAP surface receptor while providing a stable and folded domain at the N-terminus, as well as having been used for the choice of signal peptide. The monomeric luciferase used in this project, the so-called ’NanoLuc™'[17] (due to terms of simplicity referred to as ’nanoluciferase’ in the following), can be fused to other proteins in order to make their expression visible and quantifiable. This engineered luciferase emits a steady, easily detectable glow when exposed to its substrate, while being only 19 kDa large and being brighter, more specific and steadier than other luciferases commonly found in nature. Using luciferases for the quantification of expression levels offers several advantages over fluorescent proteins, including higher sensitivity and smaller size.
CMV promoter
Last but not least, high expression levels of the receptor, so that a high avidity for the interaction may be reached. The CMV promoter (taken from the parts registry, [http://parts.igem.org/Part:BBa_K74709 BBa_K74709]) therefore enables constitutively high expression of the receptor.
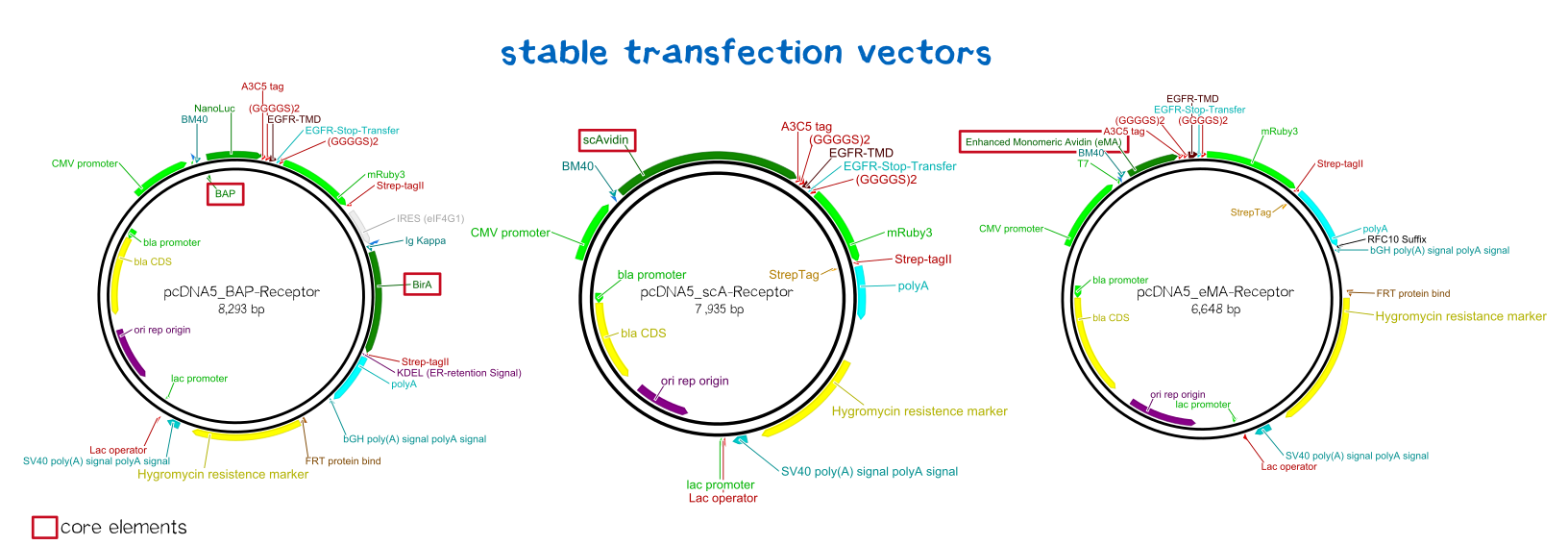
BioBrick-compatible vectors for mammalian cells: transient transfection & stable integration
Semi-stable transfection of the receptors in human cells
For the first trials of testing our constructs in human cells, we chose the MEXi™ cell line for semi-stable transfection. MEXi™ is a suspension HEK293E cell line by iba lifesciences that encodes the viral protein EBNA1 in it's genome. EBNA1, being a protein derived from Eppstein-Barr-Virus that mediates the viruses' replication initiation, gene regulation and maintenance of its DNA in the host cell. The second part of the MEXi™ system is made up by the pDSG-IBA vector that carries the specific origin of replication that is recognized by EBNA1. EBNA1 binds to this origin, and recruits the replication apparatus as well as inducing the maintenance of the inserted DNA in the host's nucleus, leading to the episomal maintenance and co-replication of the plasmid during every division cycle.[18]
Thus, we cloned our receptor constructs into the pDSG-IBA vector and then transfected the MEXi™ cells using the PEI transfection method (see methods). Polyethylene imin (PEI) as a cationic polymer forms complexes with the negatively charged DNA. These complexes are taken up by the cell via endocytosis and carried to the nucleus. [19]
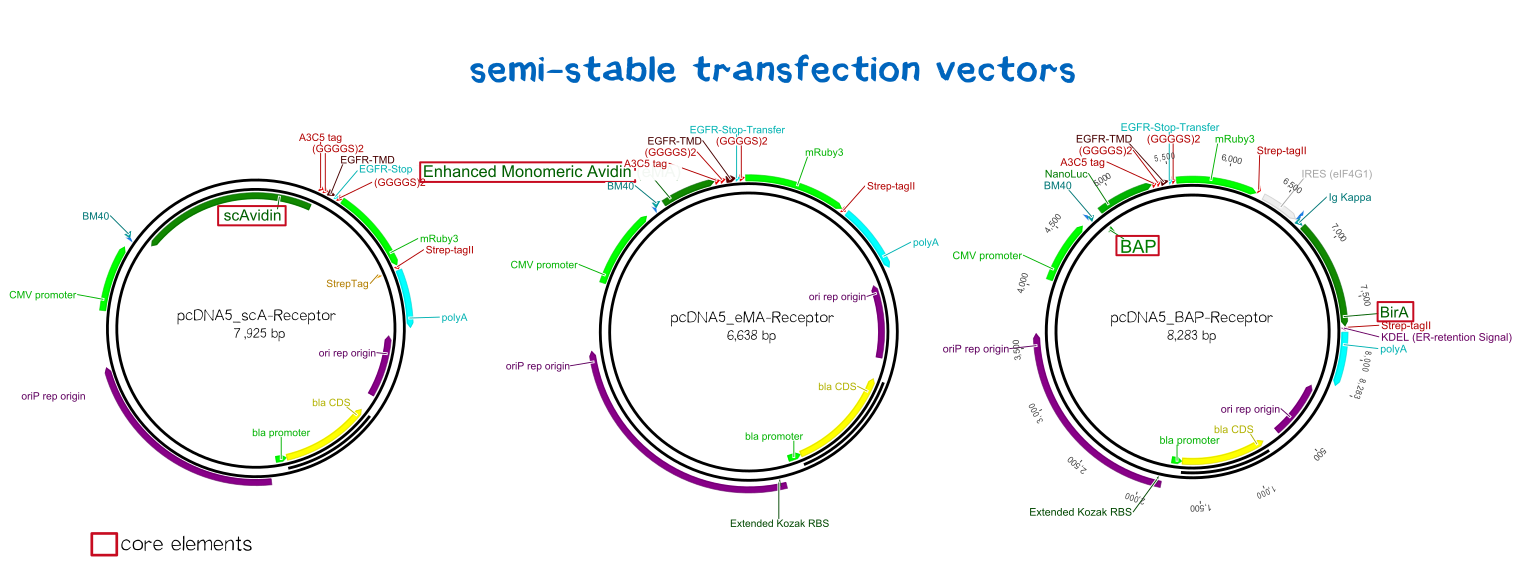
- Abbildung semistabile transfektion schematisch
An everlasting bond: creating a stable cell line
Printing tissues - or even only parts of organs - requires billions and bilions of cells. An average liver, for example, weighs about three pounds and hosts ~108 cells per gram tissue. [20] [21]
Since transiently and even semi-stably transfected cells do not propagate the transfected DNA indefinitely, repeated transfection of cells would be unwise due to the high consumption of time and ressources. Therefore, we created three stable cell lines that chromosomally encode the respective receptor constructs. For this purpose we used the FlpIn™-System by invitrogen. The three compound system consists of HEK-TRex™-293 cells that harbour one chromosomal Flp recombination target (FRT) site, a Flp recombinase expression plasmid and the plasmid encoding the receptor as well as another FRT site for stable integration.[22] The detailed principle of the intergration is described in the figure below.
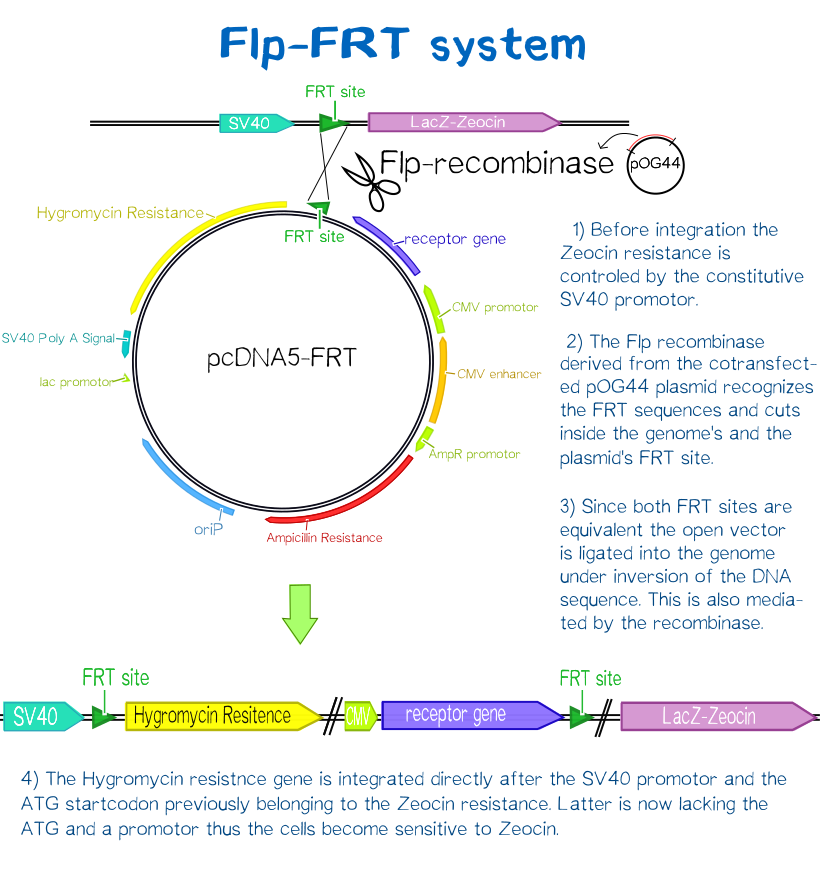
Thus the transfectant cells can be easily selected via hygromycin B. To ensure that the resistence does not come from non-intergrated plasmid, this process takes several weeks and divison cycles.
The succesful integration of the pcDNA plasmids was verified by genome-PCR (see methods). Therefor we designed three primers: One that binds to the SV40 promotor and part of the following FRT site, one that binds part the hygromycin restistance gene and another one for the lacZ-Zeocin sequence. In the event of stable integration the size of the amplicons will be around 550 bp, according to the distance between the SV40 and the hygromycine resistance gene. Wheareas in case of no intergration the amplicon size will be around 400 since this corresponds to the distance between the SV40 and the lacZ-Zeocin sequences. These results are shown in figure on the right. The lower band visible in the samples 2-5 leads us to the conslusion that despite hygromycine selection the cells are not yet a monoculture, something we also observed in the flow cytometry analysis further discussed below.
The succesful integration of the pcDNA plasmids was verified by genome-PCR (see methods). Therefor we designed three primers: One that binds to the SV40 promotor and part of the following FRT site, one that binds part the hygromycin restistance gene and another one for the lacZ-Zeocin sequence. In the event of stable integration the size of the amplicons will be around 550 bp, according to the distance between the SV40 and the hygromycine resistance gene. Wheareas in case of no intergration the amplicon size will be around 400 since this corresponds to the distance between the SV40 and the lacZ-Zeocin sequences. These results are shown in figure on the right. The lower band visible in the samples 2-5 leads us to the conslusion that despite hygromycine selection the cells are not yet a monoculture, something we also observed in the flow cytometry analysis further discussed below.
- Abbildung mit stabiler Integration, Zeocin-Test, genomische PCR
Subcellular protein localization using fluorescence microscopy and western blot
For better traceability we supplied all of our receptors with reporter genes like mRuby and/or NanoLuciferase and several tags like A3C5 and Strep-Tag.
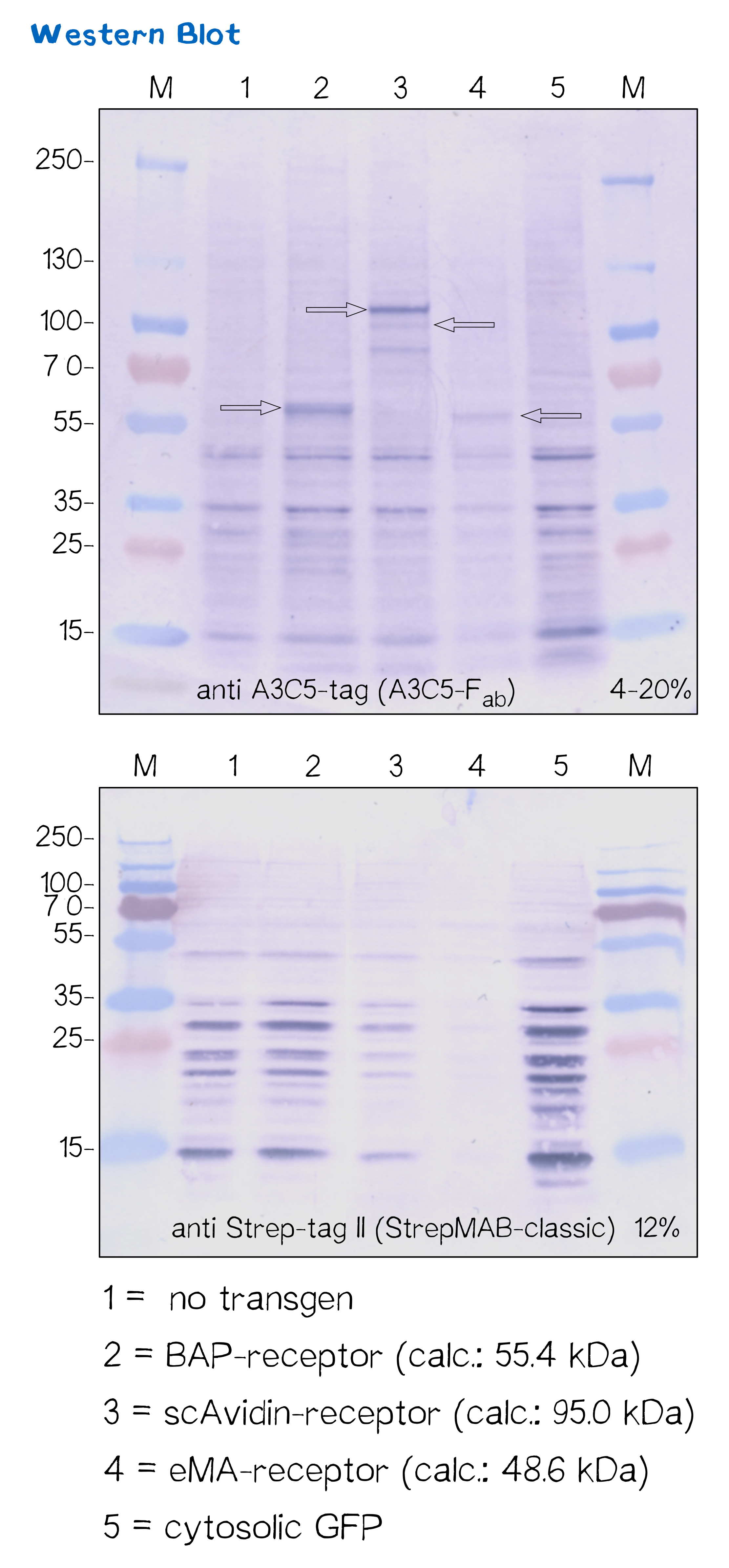
After transfecting the mammalian cells with the receptor genes and selecting them for 2 weeks on hygromycin we lysed the cells and checked for the presence of Strep- and A3C5-tag via western blot using primary antibodies against the tags und secondary alkaline phosphatase coupled antibodies (see methods). As the figure shows the Strep-tag wasn't detectable via WB however the A3C5-tag-WB shoes significant singular bands of the size of our proteins.
Quantification of functionalized membrane proteins using flow cytometry

Immunochemical detecton of functionalized membrane protein using Western Blot

In order to detect our three
| Receptor | BioBrick | Amino acids | Moelcular mass [Da] |
| BAP-Receptor | [http://parts.igem.org/Part:BBa_K2170000 BBa_K2170000] | 507 | 55 440 |
| eMA-Receptor | [http://parts.igem.org/Part:BBa_K2170001 BBa_K2170001] | 448 | 48 618 |
| scAvidin-Receptor | [http://parts.igem.org/Part:BBa_K2170002 BBa_K2170002] | 877 | 95 028 |
Quantification of mRNA expression levels by RT-q-PCR
The characterization of our transfected mammalian cells showed that the two receptors containing an avidin variant were expressed and translocated to the outer cell membrane. The biotin-presenting receptor containing the biotin-acceptor peptide, though, showed a very low fluorescence signal. Therefore, the success of the stable integration was tested by PCR of the genomic integration, which lead to the conclusion that the construct was transfected and integrated sucessfully, hinting at a problem downstream of the integration. In order to understand where the source of this problem was, we took a closer look at the expression levels of the different receptors by analyzing mRNA levels in comparison to each other and to the housekeeping gene (HGK) GAPDH.

For a better comparibility, we used the same amount of cells for each receptor and not only reviewed cells with stably integrated but also cells with the episomal stable receptor constucts. During the first step of RT-qPCR the total mRNA in converted to cDNA by a reverse transcriptase, which is then used in the actual quantitative PCR to quantify the amount of a specific mRNA was produced by the cells. For this reason two primer pairs were designed. One of them was designed to bind the exact same region in all of the different receptor mRNAs and amplify the same part of the receptor. Contrary to that the second pair was supposed to bind only in the biotin presenting receptor and amplify a part that is unique to this receptor. By this method we wanted to observe if only one part of the mRNA is unstable or if the general mRNA level of this receptor is decreased.
For the evaluation of the raw data we used the ΔΔCP method in which the crossing points (CP values) of a gene of interest and a of reference gene are compared.
In the following you can see the results of our experiments. We compared the expression levels of Trex which were transfected with our different receptor constructs and with non-transfected (NT) Trexs.
High resolution imaging using scanning electron microscopy (SEM)

{kind=link}
{kind=link}
References
- ↑ Chen, I., Howarth, M., Lin, W., & Ting, A. Y. (2005). Site-specific labeling of cell surface proteins with biophysical probes using biotin ligase. Nature methods, 2(2), 99-104.
- ↑ Rapoport, T. A. (2007). Protein translocation across the eukaryotic endoplasmic reticulum and bacterial plasma membranes. Nature, 450(7170), 663-669.
- ↑ Taken from Alberts, B., Bray, D., Hopkin, K., Johnson, A., Lewis, J., Raff, M., ... & Walter, P. (2010). Essential cell biology. Garland Science.
- ↑ Walter, P., Ibrahimi, I., & Blobel, G. U. N. T. E. R. (1981). Translocation of proteins across the endoplasmic reticulum. I. Signal recognition protein (SRP) binds to in-vitro-assembled polysomes synthesizing secretory protein. The Journal of Cell Biology, 91(2), 545-550.
- ↑ Keenan, Robert J., et al. "The signal recognition particle." Annual review of biochemistry 70.1 (2001): 755-775.
- ↑ Kozak, M. (1989). The scanning model for translation: an update. The Journal of cell biology, 108(2), 229-241.
- ↑ Kozak, M. (1987). An analysis of 5'-noncoding sequences from 699 vertebrate messenger RNAs. Nucleic acids research, 15(20), 8125-8148.
- ↑ Petersen, T. N., Brunak, S., von Heijne, G., & Nielsen, H. (2011). SignalP 4.0: discriminating signal peptides from transmembrane regions. Nature methods, 8(10), 785-786.
- ↑ Sonnhammer, E. L., Von Heijne, G., & Krogh, A. (1998, July). A hidden Markov model for predicting transmembrane helices in protein sequences. In Ismb (Vol. 6, pp. 175-182).
- ↑ Alexander, H., Harpprecht, J., Podzuweit, H. G., Rautenberg, P., & Müller-Ruchholtz, W. (1994). Human monoclonal antibodies recognize early and late viral proteins of human cytomegalovirus. Human Antibodies, 5(1-2), 81-90.
- ↑ Kredel, S., Oswald, F., Nienhaus, K., Deuschle, K., Röcker, C., Wolff, M., ... & Wiedenmann, J. (2009). mRuby, a bright monomeric red fluorescent protein for labeling of subcellular structures. PloS one, 4(2), e4391.
- ↑ Bajar, B. T., Wang, E. S., Lam, A. J., Kim, B. B., Jacobs, C. L., Howe, E. S., ... & Chu, J. (2016). Improving brightness and photostability of green and red fluorescent proteins for live cell imaging and FRET reporting. Scientific reports, 6.
- ↑ Schmidt, T. G., & Skerra, A. (2007). The Strep-tag system for one-step purification and high-affinity detection or capturing of proteins. Nature protocols, 2(6), 1528-1535.
- ↑ Case, J. F. (2004). Flight studies on photic communication by the firefly Photinus pyralis. Integrative and comparative biology, 44(3), 250-258.
- ↑ Zhang, L., Song, G., Xu, T., Wu, Q. P., Shao, X. X., Liu, Y. L., ... & Guo, Z. Y. (2013). A novel ultrasensitive bioluminescent receptor-binding assay of INSL3 through chemical conjugation with nanoluciferase. Biochimie, 95(12), 2454-2459.
- ↑ Gould, S. J., & Subramani, S. (1988). Firefly luciferase as a tool in molecular and cell biology. Analytical biochemistry, 175(1), 5-13.
- ↑ Hall, M. P., Unch, J., Binkowski, B. F., Valley, M. P., Butler, B. L., Wood, M. G., ... & Robers, M. B. (2012). Engineered luciferase reporter from a deep sea shrimp utilizing a novel imidazopyrazinone substrate. ACS chemical biology, 7(11), 1848-1857.
- ↑ Nanbo, A., Sugden, A., & Sugden, B. (2007). The coupling of synthesis and partitioning of EBV's plasmid replicon is revealed in live cells. The EMBO journal, 26(19), 4252-4262.
- ↑ Boussif, O., Lezoualc'h, F., Zanta, M. A., Mergny, M. D., Scherman, D., Demeneix, B., & Behr, J. P. (1995). A versatile vector for gene and oligonucleotide transfer into cells in culture and in vivo: polyethylenimine.Proceedings of the National Academy of Sciences, 92(16), 7297-7301.
- ↑ Molina, D. K., & DiMaio, V. J. (2012). Normal organ weights in men: part II—the brain, lungs, liver, spleen, and kidneys. The American journal of forensic medicine and pathology, 33(4), 368-372.
- ↑ Wilson, Z. E., Rostami‐Hodjegan, A., Burn, J. L., Tooley, A., Boyle, J., Ellis, S. W., & Tucker, G. T. (2003). Inter‐individual variability in levels of human microsomal protein and hepatocellularity per gram of liver. British journal of clinical pharmacology, 56(4), 433-440.
- ↑ https://www.thermofisher.com/de/de/home/references/protocols/proteins-expression-isolation-and-analysis/protein-expression-protocol/flp-in-system-for-generating-constitutive-expression-cell-lines.html#prot4
- ↑ Blochlinger, K. A. R. E. N., & Diggelmann, H. E. I. D. I. (1984). Hygromycin B phosphotransferase as a selectable marker for DNA transfer experiments with higher eucaryotic cells. Molecular and cellular biology, 4(12), 2929-2931.