Team:Aalto-Helsinki/Description
PROJECT
Introduction
Right from the beginning our team wanted to address a real-world issue which was local in Finland and didn’t have any current solutions available. In one of our brainstorming sessions we came up with the problem of blue-green algae. Check out our introduction video “Matti and the cyanobacteria” to see how our solution for this problem would work!
Project overview
Our project can be divided into two parts: firstly, detecting the cyanobacterial toxin microcystin from water and secondly, degrading it when necessary. Both the detection and degradation happen with the help of modified baker’s yeast, Saccharomyces cerevisiae . Our idea is summed up in Figure 1.
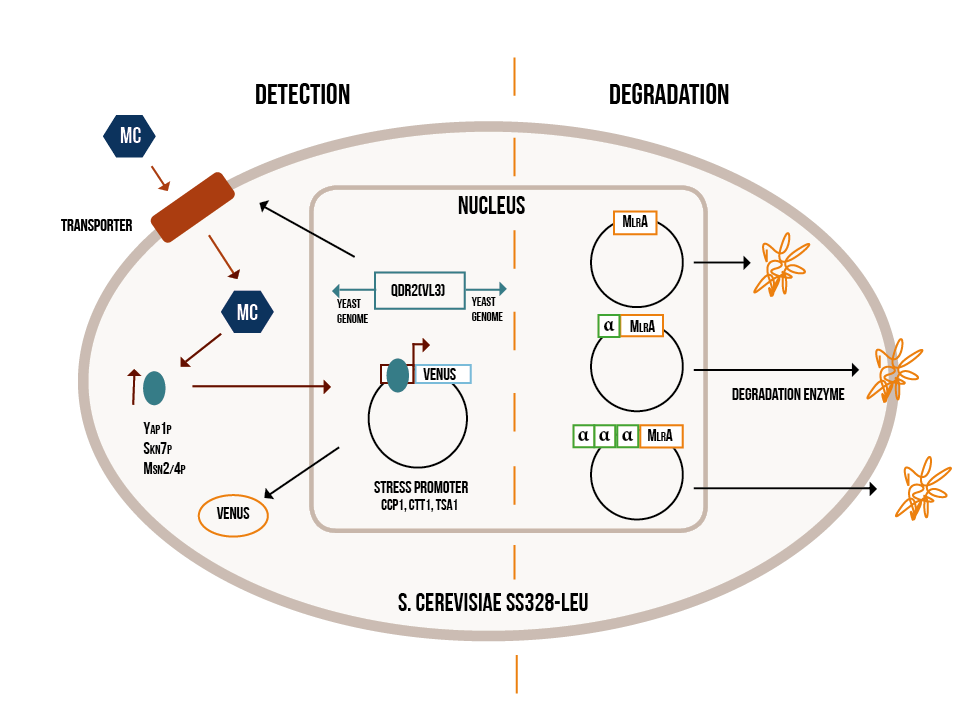
Detection:
The first step in the detection of microcystin is to get the toxin inside the yeast cell. Normal yeast lab strains are not known to be capable of doing this, but there is one strain, normally used in winemaking, which is capable of importing microcystins. This strain (VL3) has a variant of a transporter normally present in yeast that is capable of importing the toxin (Valério, 2014). In our project, we will replace our strain’s original, non-toxin importing transporter gene from the yeast’s genome with the one from VL3. This way our lab strain will also be able to transport the microcystin inside the cell.
When microcystin is imported into eukaryotic cells, it causes oxidative stress. This increases the activity of certain transcription factors, which activate genes involved in the oxidative stress response (He and Fassler, 2005). We take advantage of this natural response in our detection mechanism. In our engineered yeast, promoter regions of genes involved in oxidative stress response are coupled to genes coding for yellow fluorescent protein. Consequently, when encountering microcystin, our yeast will start to produce yellow fluorescent protein, and the resulting fluorescence can then be measured. We hope that the level of fluorescence will correspond to the extent of oxidative stress, and therefore also the concentration of microcystin.
The detection mechanism will be modelled to support our experiments and to provide suggestions for actual promoter design. We also hope to be able to predict the response of our promoter constructs to different microcystin levels with the help of modeling results.
Degradation:
In the degradation part of our project, we introduce a gene coding for an enzyme called microcystinase (MlrA) into yeast cells. Microcystinase is produced in certain gram-negative bacteria and it linearizes microcystins, making them significantly less toxic (Bourne, 1996). We will have three variants of the enzyme to optimize its expression in yeast cells. The hypothesized localisation of the variants are shown in Figure 1.
“ Both the detection and degradation are implemented with the help of modified baker’s yeast, Saccharomyces cerevisiae ”
Background
Cyanobacteria and microcystin-LR
In late summers every year, blooms of blue-green algae start to appear in lakes in many places over the world, including in Finland. Blue-green algae are photosynthetic, nitrogen fixing bacteria that live in waters and soil. Especially in the late summer, the eutrophication of lakes provides perfect conditions for cyanobacteria to reproduce exponentially. Even though cyanobacteria are important for the ecosystem because they produce oxygen and fix nitrogen from the atmosphere, they also pose health risks to humans and animals, as well as problems for water industries.
Cyanobacteria can cause skin irritation, nausea, headache, vomiting and even death (WHO, 2003). Cases of human death caused by water contaminated with cyanobacteria have been reported; for example in 1996, over 50 haemolysis patients died due to cyanobacterial toxin contaminated dialysis water (Welgamage, 2012). Each year in Finland, pets die from drinking water contaminated by cyanobacteria. The cyanobacteria also pose a problem for water purification plants, since they need to get rid of any possible contaminants in the water. This also results in significant costs, as plants have to have proper filtration systems. In 2014, around half a million people were left without pure water for two days as a water purification plant in the US was contaminated with cyanobacteria (Plumer, 2014).
These health risks and problems are mainly due to the toxins produced by the cyanobacteria. These toxins, also known as cyanotoxins, can be classified as neurotoxins, cytotoxins, dermatoxins, endotoxins and hepatotoxins.
There are two different kind of hepatotoxins: microcystins and nodularin. Both are cyclic peptides that have several shared features. One of these features is that both have the unique amino acid Adda incorporated into their structure; Adda is only found in cyanobacteria. Microcystins are normally found in fresh waters, while nodularin is encountered in brackish waters. As there is more previous research on microcystins, and they are the most common cyanotoxins in freshwater systems, our research has focused on it. (Welgamage, 2012)
Microcystins are cyclic cyanotoxins that consist of seven amino acids. They have the unique β-amino acid Adda, and two of the seven amino acids vary between different variants of microcystin. Due to the variation in these two amino acids, there are at least a hundred different variants of microcystin. The most common variant is microcystin-LR (MC-LR), which has leucine and arginine as the varying amino acids. It is also the most potent of all microcystins. (Welgamage, 2012) The structure of microcystin-LR is presented in the figure 2.
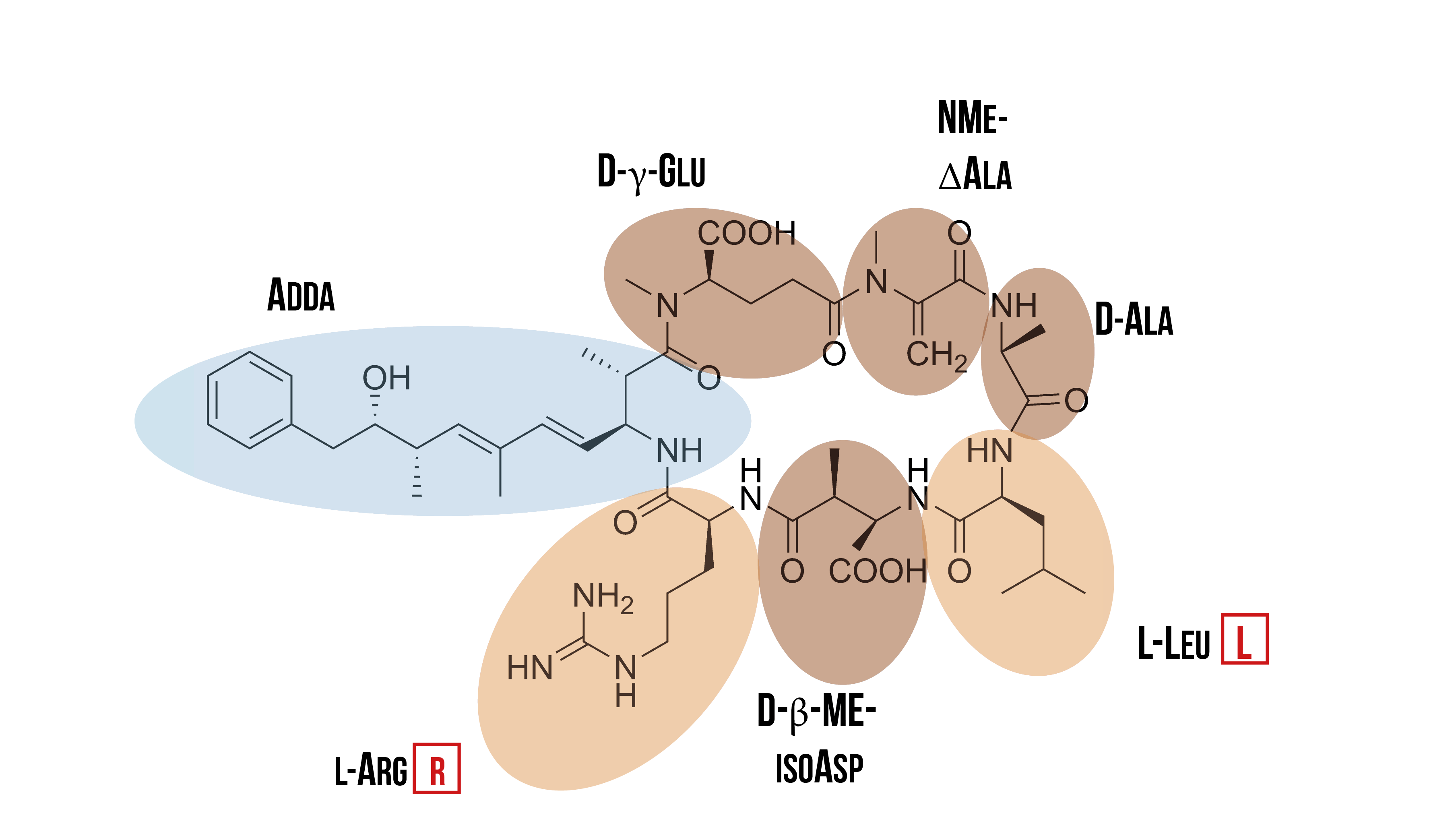
Out of all cyanotoxins, the ones that have a cyclic peptide structure (such as MC-LR) are the most dangerous to humans. Because of this cyclic structure, they are very stable, to the extent that they do not get degraded in the acidic environment of the stomach upon ingestion. Because of this, they can reach the gastrointestinal tract and the liver, where they inflict their toxic effects. High dosages can even cause death, with the LD 50 value being 5 mg/kg in mice. Exposure to lower dosages may also promote tumor formation. (WHO, 2003)
Different species of cyanobacteria produce different toxins, but there can be some variation between different strains of a given species. It is also possible that some cyanobacteria produce toxins while other individuals from the same strain do not produce any at all. In addition, the cyanotoxins are released only as the cells die and lyse. (WHO, 2003) This makes it difficult to quantify the toxin concentration in the water. According experts from SYKE (Finnish Environment Institute), who we interviewed, at the moment there is no affordable, easy way to measure the microcystin concentrations in waters. Detection of algal blooms is done mainly by satellite images or indirectly by measuring other factors such as phosphorous concentrations, which give a rough estimate of the amount of cyanobacteria. Other ways to detect the cyanobacteria are for example optical sensors, cameras, DNA analyses, chlorophyll analyses and fingerprint matrixes. These methods, however, do not give any indication of the amount of microcystin or other cyanotoxins. Methods for measuring the concentration of cyanotoxins require laboratory facilities, and are based on antibody detection.
For the degradation of microcystin, there are no existing methods. In the bigger picture, microcystins and other cyanotoxins are a result of eutrophication, as that is the reason for cyanobacterial blooms which then lead to the production of the toxins. Therefore, to permanently reduce the problem of cyanotoxins, the causes of eutrophication must be controlled. Examples of measures that have helped in the prevention of eutrophication include restrictions on the use of different fertilizers and preventing factories from letting their waste waters get to natural water systems. Even though algal blooms have reduced over the years, in many places the toxin concentrations still rise above the safety limit every summer. This is why we want to provide an easy and affordable way to detect and degrade microcystins.
Why we are using yeast
The yeast Saccharomyces cerevisiae is a unicellular eukaryote, and as a such, it has certain advantages compared to e.g. Escherichia coli . It has several benefits that make it an easy chassis to work with, such as a fully sequenced genome, fast growth, and available protocols. However, as an eukaryote it has the additional advantage of sharing many similar cellular functions with higher eukaryotes.
In iGEM, S. cerevisiae is not a popular chassis organism. This is easy to understand as yeast grows slower than bacteria, and with the limited time in the competition it is no wonder that many teams opt for a faster growing chassis. It is also likely that students have more experience in working with E. coli , and as a result, they favour a bacterial chassis. Moreover, in many projects working with an eukaryotic chassis wouldn’t bring any notable benefits. This preference for prokaryotic hosts can clearly be seen in the BioBrick Registry: there are hardly any yeast-compatible BioBricks, and most of them are poorly characterized and not in stock.
The main reason we chose to use yeast is that it has many similar cellular functions compared to humans. As we researched the toxicity effects microcystin has on mammalian cells, we discovered that the toxin has the same basic toxicity mechanism also in yeast cells, but it has no notable effect on e.g. E. coli (Yang, 2008). Also, with our project, we want to make synthetic biology more acceptable to people, and bring a change in the public views surrounding it. Using S. cerevisiae , or baker’s or brewer’s yeast as many outside the scientific community call it, as a chassis could help in this. By using a familiar organism with neutral connotations as a chassis, we can show that synthetic biology doesn’t have to be something scary or foreign. Additionally, because our detection mechanism is engineered in yeast, it was logical to implement the degradation part in yeast, too. That way, it will be possible to couple these two together in the same organism if desired.
In addition, if enzyme production in S. cerevisiae is successful, this could open possibilities for attempting to produce the enzyme in other eukaryotic organisms capable of even higher production yields, such as the yeast Pichia pastoris (Ahmad et al. 2014). This could enable more efficient mass production of the enzyme. An additional benefit of yeast is that it has an efficient secretion mechanism (Bitter, 1984); this would greatly facilitate mass production and purification of our enzyme, if it is possible to secrete it.
Our team was also lucky to work in a laboratory where people work routinely with yeast; we could ask help whenever we needed it. The advice we received from people working with yeast made our work a lot easier and taught us much. However, this was not the case for every team working with yeast this year. One of our collaborations was in fact helping the Uppsala team by giving them protocols for working with yeast and also some advice we had received from our lab, since the Uppsala team did not have anyone at their institution who could have helped them.
“ This is why we want to provide an easy and affordable way to detect and degrade microcystins ”
Detection
Background
When studying the toxicity of the cyanobacterial toxin microcystin-LR in higher eukaryotic cells, we found out that it causes oxidative stress in them. This is due to inhibition of protein phosphatases 1 and 2A (PP1 and PP2A) which then leads to hyperphosphorylation of target proteins. This excessive phosphorylation in turn increases the formation of reactive oxygen species (ROS), resulting in a oxidative stress response. (Campos and Vasconcelos, 2010) This is presented in the figure 3. Reactive oxygen species have many effects in the cell. For example, catalase production is significantly increased in the presence of ROS. Catalase is an enzyme found in nearly all living organisms that catalyses the decomposition of hydrogen peroxide. It is one of the most important enzymes in protecting the cell from reactive oxygen species meaning that its production is activated when there is oxidative stress.

We found out that similar toxicity effects can be observed in some lower eukaryotes, such as the yeast Saccharomyces cerevisiae . (Valério et al., 2014, Valério et al., 2016) Realizing this made us think of how we could use MC-LR's natural toxicity effects to sense the toxin concentrations.
Stress Promoters
Various articles (Estruch, 2000, He et al., 2005) indicated that oxidative stress increases the activity of certain transcription factors in S. cerevisiae cells. Two transcription factors activated by oxidative stress are Skn7p and Yap1p. These transcription factors have specific binding sites in the promoter areas of genes that respond to oxidative stress. Skn7p binds to OSRE (oxidative stress response element) and Yap1p binds to YRE (Yap1p response element). Some of the best characterized promoter regions that these transcription factors bind to are those for the genes TSA1 (thioredoxin peroxidase) and CCP1 (mitochondrial cytochrome-c peroxidase) (He et al. 2005). The gene that was reported to have highest response against microcystin was CTT1 (cytosolic catalase T) (Valério et al., 2014). We chose to look into these three promoters more deeply, and this led us to the idea of creating a sensor by fusing one of these promoter areas with fluorescent protein production and implementing this in S. cerevisiae .
This sensor would produce a fluorescent signal when the activation of the transcription factors Skn7p and Yap1p has been induced – in other words, when the S. cerevisiae cells encounter stress. As microcystin is a rather effective stress-generating factor, and as most sources of oxidative stress (such as xenobiotics and heavy metals) are rare in nature (Farrugia et al., 2012), we can assume that it is the main stress producing factor found in Finnish lakes. This leads to our hypothesis: fluorescent colour produced by our engineered S. cerevisiae is proportional to microcystin concentration in measured water samples.
Transporter
An important prerequisite for the function of the stress promoters is that the toxin must be imported into the cell. Microcystin-LR doesn’t pass the cell membrane without the help of specific transporter proteins. In human liver cells, the membrane bound OATP transporter (organic anion transport peptide) was identified to imports MCs into the cell. OATPs belong to the superfamily of solute carriers and mediate the uptake of endogenous compounds such as hormones or bile salts but also a variety of xenobiotics (Faltermann et al., 2016). In yeast, the QDR2 transporter has a similar function. It has a broad substrate specificity and can transport many mono- and divalent cations as well as a variety or drugs. According to Valerio et al., the QDR2 transporter from the yeast strain VL3 is the most similar to human OATP out of all S. cerevisiae strains.We also did a BLAST search to confirm this data and come to the conclusion that QDR2 transporter from VL3 corresponded best to the human OATP transporter out of all yeast transporters. Transporters from all other common S. cerevisiae strains differ significantly from these.
It has been confirmed that the QDR2 transporter from VL3 can import microcystin (Valério et al., 2014). We reasoned that by replacing the original QDR2 transporter of a S. cerevisiae lab strain (W303a/SS238) with the VL3 variant of the QDR2 transporter we could enable toxin import in a new strain. The reason for not wanting to work with VL3 was that it’s used and optimized for wine production, not for extensive gene engineering. Another reason for not choosing to engineer VL3 was that we would have had to do auxotrophic mutations for it first as such kind of this strain was not available. However, we planned to use VL3 as a positive control in our experiments to compare how changing the transporter of a lab strain effects observed toxicity effects. This way, we hope to see whether toxicity behaviour corresponding to VL3 can be observed.
Final Product
For the final product, we will need to have both the VL3 transporter and the stress promoter in yeast cells. The colour of two lake water samples, one supplemented with a defined quantity of engineered S. cerevisiae cells and one with unmodified yeast, can then be compared, and the toxin concentration can be calculated easily from a standard curve utilizing known toxin concentrations.
In the final product, we would have a test device containing the measurement and control yeasts. The user could add a small amount of sample water to be measured and analyse it with a mobile app that tells about the toxicity based on the colour difference between the sample and the control. The idea is explained shortly in figure 4.
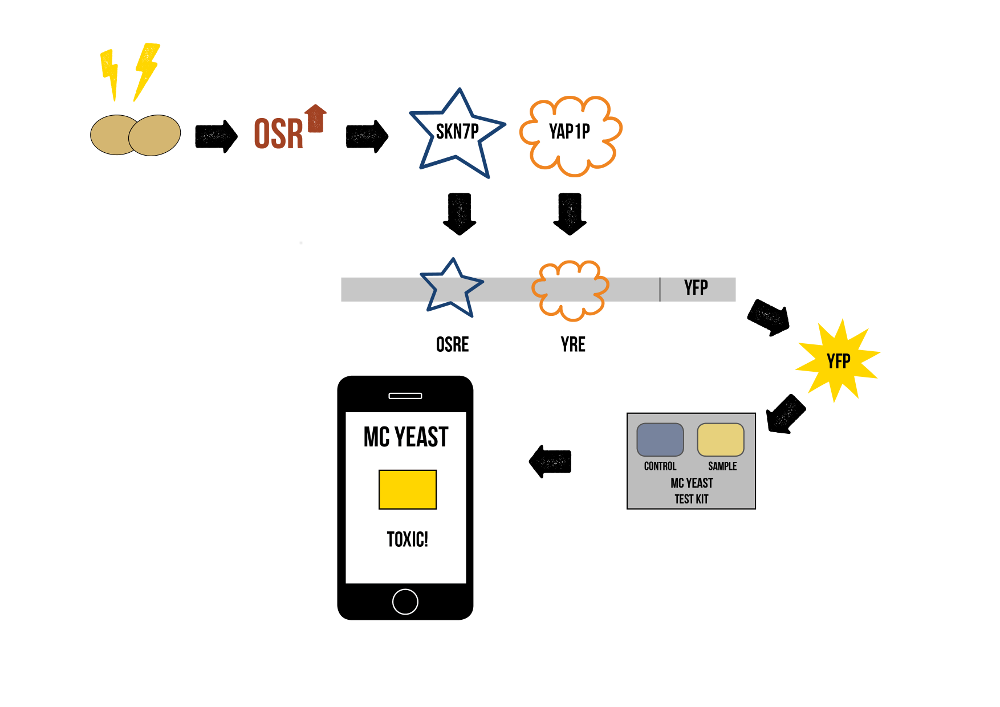
Design
Promoters
We chose three whose transcription is regulated greatly by oxidative stress or which promoters are well characterized. These genes were TSA1 (Saccharomyces genome database ID: S000004490), CTT1 (SGD ID: S000003320) and CCP1 (SGD ID: S000001774). We decided to incorporate the promoters of these genes directly into our stress promoter constructs. Binding sites for transcription factors of our interest are identified the region of 1-300 bp before the start codon, but the length of the whole promoter area has not been defined. As promoter areas can be up to 1000 bp long, we decided to use upstream sequences longer than 300 bp. For TSA1 we decided take promoter region of 260 base pairs, as that was the longest we could take so that it didn’t overlap with the previous gene in yeast genome. In the case of CCP1, we decided to have a promoter region of 756 bp, as that was the same that had been used and proved to be working in our reference article (He et al., 2005). Unlike for CCP1 and TSA1, we didn’t find as clear an explanation for the length of CTT1. We only found that the reference article had done its experiment with a 479 bp upstream sequence so we decided to be sure that it was long enough and took 924 bp.
For our synthesized gene constructs, these promoters were then directly followed by the coding region of a yellow fluorescent protein. This construct was designed to replace the original promoter and multiple cloning site of our yeast plasmid. This way we could make YFP production dependent on oxidative stress. Venus yellow fluorescent protein was chosen as the visual reporter as it was supposed to be bright, stable in a variety of conditions (Shaner et al., 2015) and the sequence of codon optimized version for S. cerevisiae was available. Besides promoter + venus constructs, we decided to expressed Venus under control of one constitutive and one inducible promoter to get more reliable information about expression of Venus in yeast, and to be able to study the effect of different conditions on its expression under known promoters. The used promoters were the galactose inducible GAL1 promoter and the strong, constitutive GPD1 promoter. These constructs will serve as a control in our stress promoter tests.
The constructs were designed so that they were compatible with both Gibson assembly and yeast recombination cloning. This means that a sequence of 30 bp that is homologous with the desired cloning location from the plasmid backbone was added to each end of the promoter + Venus construct. As a plasmid backbone, we used pRS415-GAL/GPD (pRS415 plasmid with either GPD1 or GAL1 promoters), which was already available in our lab, and suitable for protein expression in S. cerevisiae . A simplified picture of all constructs is presented in figure 5. Inserts containing homologous sites for ligation, designed promoter, and venus, were ordered from IDT.
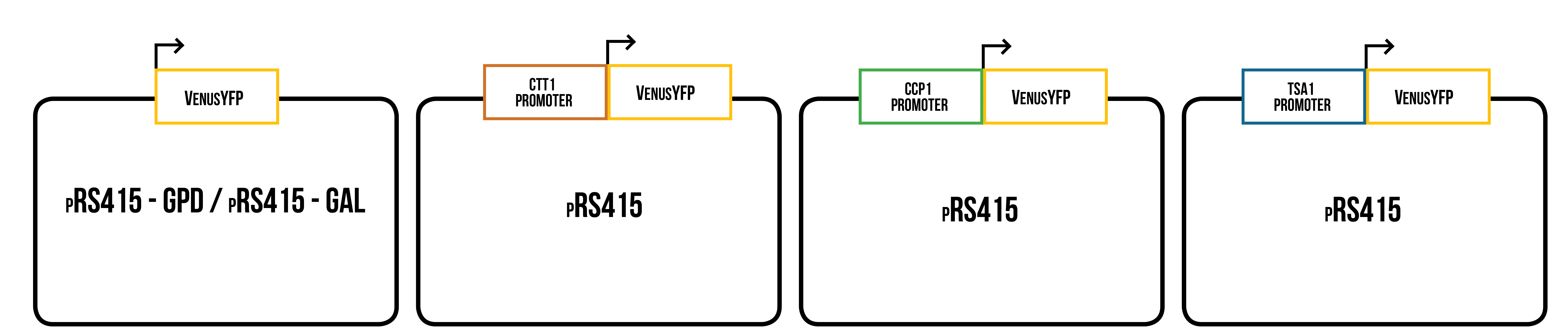
Our designed inserts are not compatible with Biobrick assembly standard 10. The reason for this was that there were no plasmid backbones or hardly any other parts designed for S. cerevisiae in stock in the iGEM registry. Additionally, the characterization of the parts were lacking. The plasmid backbone we eventually chose was pRS415-GAL, as it was easily available to us, suitable for our purpose, and there was experience that it had previously worked well in S. cerevisiae . Our plan was to modify the best of our designed inserts to be compatible with Biobrick standard 10 (to have defined prefix and suffix) after testing and analyzing them.
Our plan to test the functionality of stress promoters is to use hydrogen peroxide to induce stress in yeast cells containing stress promoter plasmids, and measure the differences in YFP fluorescence intensities. From these results, we hope to obtain a linear relation between inducing hydrogen peroxide concentration and YFP intensity. These tests cannot be done with microcystin-LR, since it isn’t imported into the cell without the VL3 transporter, and thus doesn’t cause a strong enough response. All the same, as hydrogen peroxide is the main reactive oxygen species produced in stress caused by microcystin-LR in yeast cells, we planned to use this compound directly in our primary experiments. After proving that our sensor works with hydrogen peroxide, and finishing transporter implementation, we can then transform our stress promoter into cells with the VL3 transporter already integrated into their genome. This way, we could test our promoters with microcystin.
Modelling
We are going study our detection mechanism mathematically in order to obtain better understanding of it. Our plan is to create simplified molecular and mathematical models of our detection mechanism which will describe how the transcription factors are affected and how they affect the regulation of our promoters under oxidative stress. Then, we will simulate these models with COPASI modelling software. We hope to validate our model with experimental results for each construct. In addition, we hope to determine theoretical detection thresholds and measurement accuracy estimates, as well as provide suggestions for optimal promoter design for future iterations of the project.
As modeling goes into great detail on subjects not as tightly related to the biological design of the project, it is presented in more detail in it’s own page .
Transporter
The QDR2 transporter sequences of VL3 strain were available online, but there were two nucleotides in the sequence that were not determined. The first one (nucleotide 475) was marked with M, meaning that it could be either adenine (A) or cytosine (C), and the other one (nucleotide 1126) with Y, meaning that it was either tyrosine (T) or cytosine (C). For the first nucleotide, the different bases would result in a changed amino acid at position 159 - either a leucine or isoleucine. We decided to test both transporter variants. For the second unclear base, replacing Y with T would have produced a premature stop codon at the place of amino acid 376. Because of this, we chose C, which resulted in having glutamine as amino acid 376. After this reasoning we had two different transporter variants. The following figure 6 presents the possible options we had.
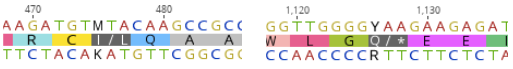
We planned to replace the transporter directly in our S. cerevisiae lab strain’s genome with homologous recombination. To be able to detect the correct expression of the transporter replacement in the eventuality that functional testing with response to MC wouldn’t produce clear enough results, we also designed constructs with strep-tag markers on both termini and a construct with a mCherry red fluorescent protein tag on the C-terminus. All designed QDR2 variants are presented in figure 7.

All inserts were designed to be amplified in the pUG6 plasmid backbone, which is commonly used to generate deletion cassettes to be used in yeast homologous recombination (Güldener et al., 1996). The pUG6 backbone contains ampicillin resistance for amplification in E. coli , and a KanMX resistance cassette for antibiotic selection in yeast, flanked by loxP sites to enable recovery of the selection marker after selection. The inserts were designed to be compatible with either Gibson assembly or yeast recombination with a linearized pUG6 plasmid backbone. The disruption cassette for genome integration can then be generated from the assembled transporter-pUG6 plasmid via PCR. The PCR is designed to generate a linear fragment containing both the transporter, the KanMX selection marker, and 50 bp long regions homologous to the intended site of recombination in the genome. (During this PCR, 50 bp long homology will be added to the 3’ end to enable genome integration. 5’ homology is designed already in the ordered insert.) This linear cassette can be then transformed into our lab strain, where it will integrate into the location of original transporter. The planned integration is illustrated in figure 8.
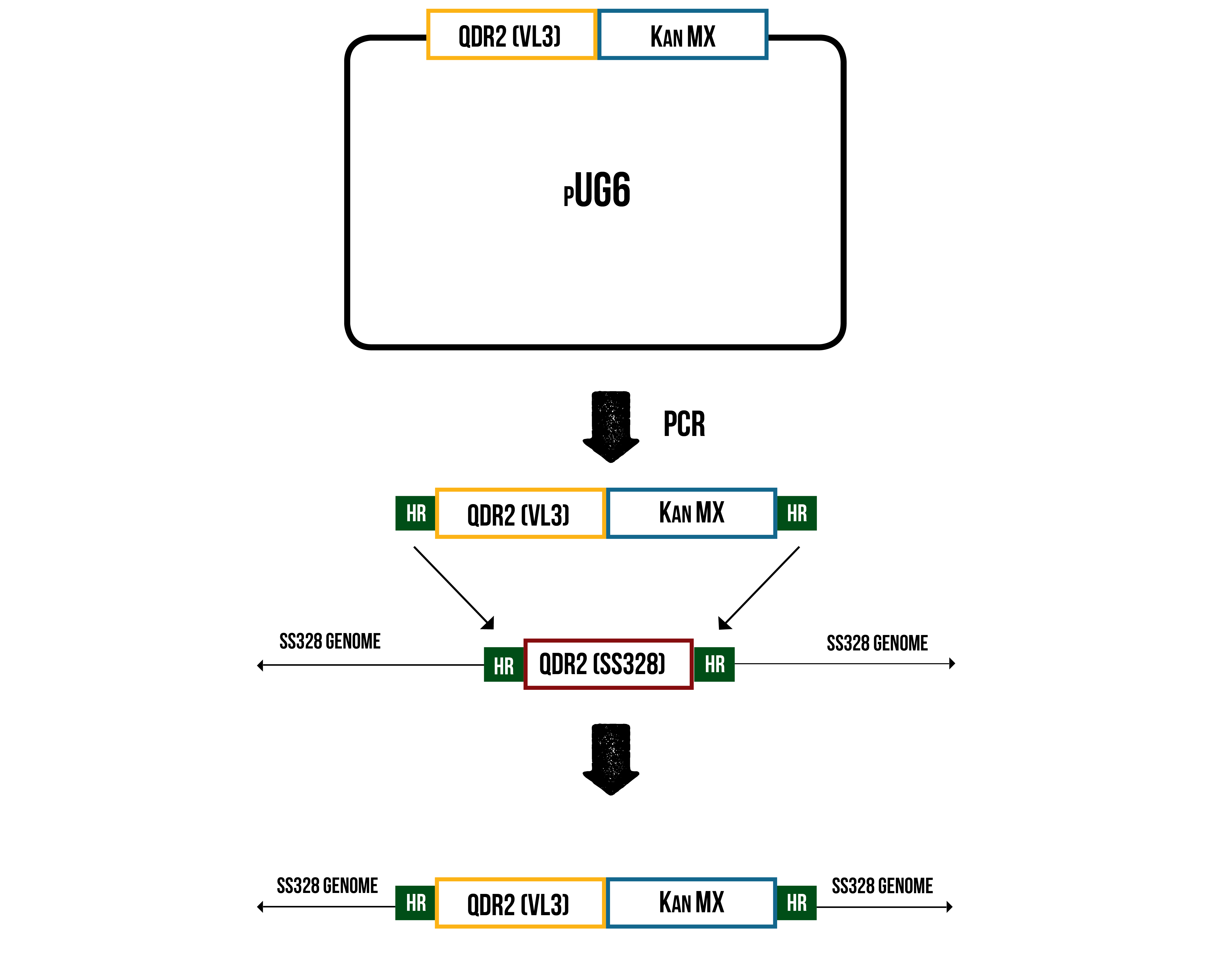
For the same reason as in the case of stress promoters, these inserts aren’t compatible with Biobrick assembly standard 10.
“ For the final product, we will need to have both the VL3 transporter and the stress promoter in yeast cells ”
Degradation
Background
The second part of our project is the degradation of microcystin, a cyanobacterial toxin, and more specifically the degradation of microcystin-LR. A naturally occurring enzyme called microcystinase (MlrA) is capable of degrading different microcystins. Our goal is to introduce this enzyme into yeast cells and optimize its expression and purification so that it can be obtained in an active form.
Microcystinase
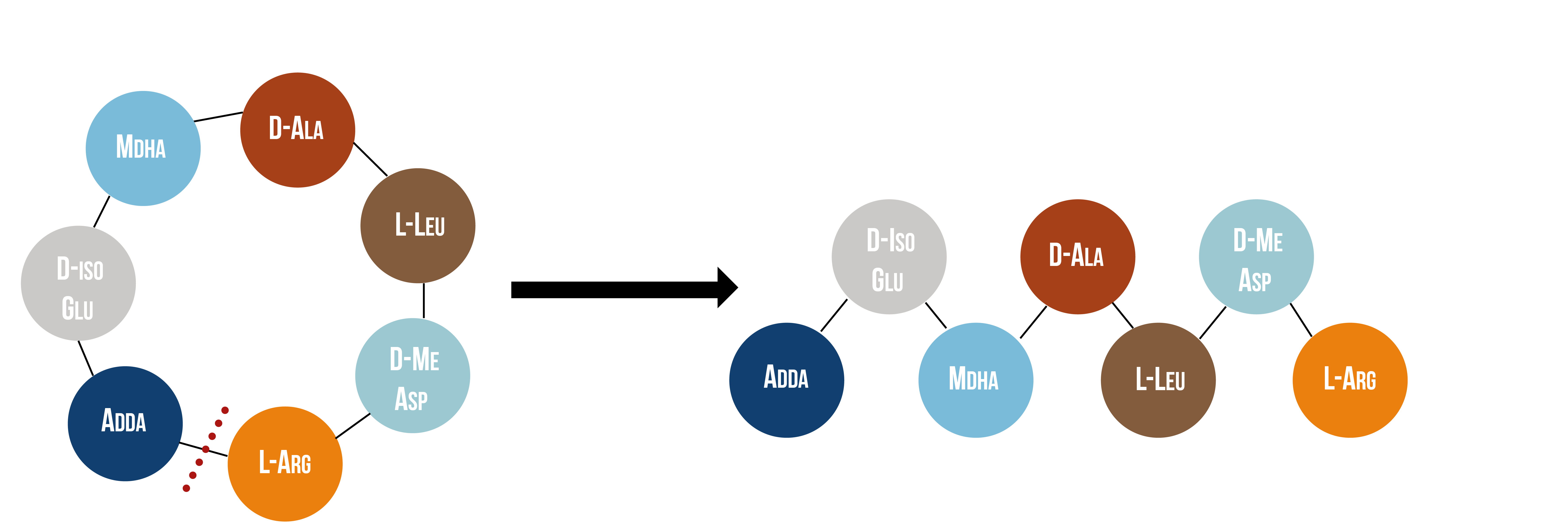
Microcystinase, or MlrA, is a member of a set of enzymes that degrade microcystins. These enzymes - MlrA, MlrB, MlrC and MlrD - are found naturally in some gram-negative bacteria, for example Sphingomonas sp. They each have a different function in the overall process of microcystin degradation. MlrA is first in the sequence to act. It hydrolyzes the peptide bond between the Adda and arginine amino acids in microcystin-LR (MC-LR). The breakage of this bond linearizes the toxin (figure 9). Following this, MlrB (a serine protease) and MlrC (a metalloprotease) are able to further degrade the linearized product into individual amino acids, which are then used in the cell’s metabolism. Therefore, the toxin is likely used as a carbon source. MlrD’s function is not fully understood at this point, but it may have a role in importing the toxins. (Bourne et al., 2001)
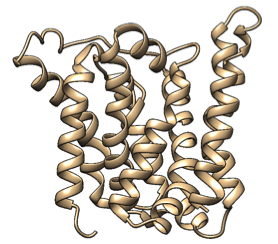
Based on a homologous structure model (figure 10) we obtained from the Phyre2 tool (Kelley et al., 2015), it seems that the enzyme is a transmembrane protein consisting of eight hydrophobic α-helices that form a channel, with the presumed active site (Bourne et al., 2012) residing in the middle. It is worth pointing out, however, that the model does not cover the whole length of the MlrA sequence, as only very few homologs were found for the sequence - the homology model is mostly based on two homologs that didn’t cover the very ends of either terminus. Other than this evidence, there are previous studies pointing to the direction of MlrA being a membrane protein (Pei et al., 2011). However, when the enzyme was heterologously produced in E. coli , it was noticed that it had localised mostly in the soluble fraction of the cytosol, in an active state (Dziga et al., 2012).
The mechanism of MlrA’s function is not yet fully understood. The homologous structure model gives some suggestions as to how the enzyme might work: it seems that one end of the channel is wider than the other, and that the active site resides in the middle of it. Consequently, if the enzyme is located on the outer membrane of the cells, it might actually be a transporter which the cells use to import the toxin and simultaneously linearize it.
It has been reported that the linearization of MC-LR renders the toxin 160 times less toxic (Bourne et al., 1996). When the blue-green algae problem is at its worst, the levels of microcystin can go up to 24 mg/L (Chorus and Bartram, 1999). However, such levels are only observed in scums, which are formed by certain cyanobacterial species such as Microcystis spp. and Anabaena spp. Our enzyme will not be able to detoxify such high toxin concentrations - a concentration of 24 mg/L would be detoxified to toxicity value corresponding to a concentration of 150 μg/L of MC-LR, which is greater than the safety limit of 1 ug/L designated by the WHO (WHO, 2003). However, scums are very conspicuous on the surface of the water, and people would instinctively not use such water for any purpose, including for sauna. The normal levels of microcystin when the cyanobacteria don’t form scum are maximally 20 μg/L (Chorus and Bartram, 1999), which can be detoxified to safe levels with MlrA.
Project Design
When designing our constructs and experiments, there were several factors we needed to consider: large-scale production of the enzyme, the enzyme being a putative membrane protein, and ensuring that the microcystin inside intact cyanobacterial cells would also be degraded.
Dziga et al. (2012) had succeeded in producing active, soluble MlrA in E. coli , but because we want our final product to be manufactured in a large scale, it seemed like there would be better ways to achieve this efficiently. Optimally, this would mean using a chassis organism capable of higher protein yields with the produced enzyme being secreted into the growth medium, since that would be the most cost-effective way of purifying it. The enzyme should also have some properties that make it easy to purify.
In the detection part of our project, we had already decided to use S. cerevisiae as our chassis. Having both parts of the project in the same chassis organism would allow us to potentially couple the two in the future. In addition, because of our considerations of enzyme mass production, a chassis capable of higher protein yields and better secretion efficiency than E. coli would be beneficial. E.g. the yeast Pichia pastoris is known for its capability to achieve extremely high cell densities and protein production yields, which is optimal when thinking of mass production (Ahmad et al., 2014). Coupling this with secretion would be ideal. Verifying that the enzyme production works in S. cerevisiae would be promising for considering potential production in eukaryotic organisms such as P. pastoris. This gave us additional motivation to use S. cerevisiae as the chassis in our project.
If the enzyme is a membrane protein as suspected, secreting it would likely not be possible. However, as it had been previously expressed in a soluble, active form in the cytoplasm of E. coli , and there is no certainty of the structure, we thought secretion might be worth attempting. A widely used secretion tag in S. cerevisiae is the mating factor α (UniProt ID: P01149), which is involved in the signaling between two yeast cells prior to conjugation. It is composed of several domains, each having different functions (figure 9). The first domain is the signal sequence, which directs the protein into the inner membranes for modifications. The second domain is the propeptide sequence for the secretion of the protein, which ends in signals for proteolytic cleavage. The propeptide is followed by the actual mating factor α sequences, each separated by spacer peptides that contain proteolytic processing signals. (Kurjan and Herskowitz, 1982)
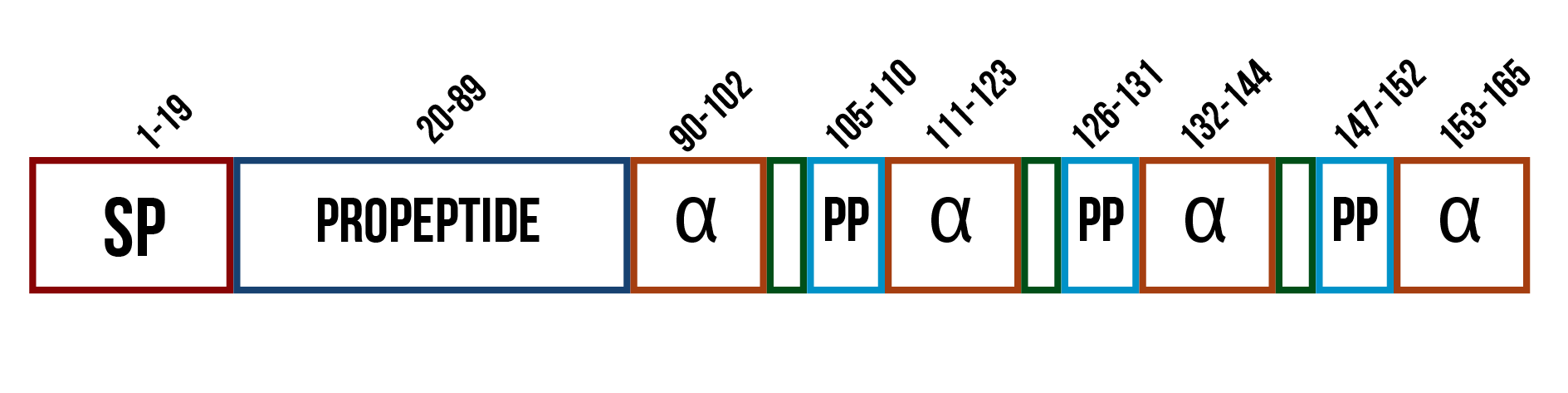
When the mating factor α is used as a secretion tag, only the first two domains are used. The tag, composed of these two parts, is added in front of the protein sequence and it guides the protein out of the cell. The mating factor α is also used as a localization tag for membrane proteins. Membrane proteins usually have hydrophobic regions, and as the mating factor α tries to secrete them, these hydrophobic regions get stuck in the plasma membrane. However, placing the signal sequence in front of the actual enzyme coding sequence might also cause some problems with correct folding of the protein or decrease the enzyme’s activity.
Because there was contradictory evidence relating to MlrA being a membrane protein, using the mating factor α served a dual purpose; if the enzyme is a membrane protein, as suggested by homology modeling, localizing it to the plasma membrane might allow for better activity against microcystin. The membranes could then possibly be purified along with the enzymes. On the other hand, if the enzyme isn’t a membrane protein, as previous reports of its expression might suggest, the mating factor α would allow for its secretion.
In order to find the conditions where the enzyme is produced best, we designed three different variants of the enzyme, all codon optimized for yeast. One is only the enzyme (figure 12), the second one has the secretion tag in the N-terminus of the enzyme (figure 13) while the third one also has the secretion tag, but instead of just one propeptide part, there are three propeptide parts (figure 14). Our hypothesis is that the first one would remain in the cytosol, the second one would be optimal for localization in the plasma membrane, and the third one would favor secretion.
We also wanted to be able to purify our produced enzymes in order to quantify the amounts produced. We chose to add a 6X His tag, which is composed of six histidine residues, in the C-terminus of our enzymes. This should make it possible for a simple purification using immobilized metal affinity chromatography (IMAC) while also allowing for detection in Western blots with anti-His tag antibodies. The His tag had also been used by Dziga et al. (2012) when expressing MlrA in E. coli , which indicated that a His tag should not interfere with the proper function of MlrA.

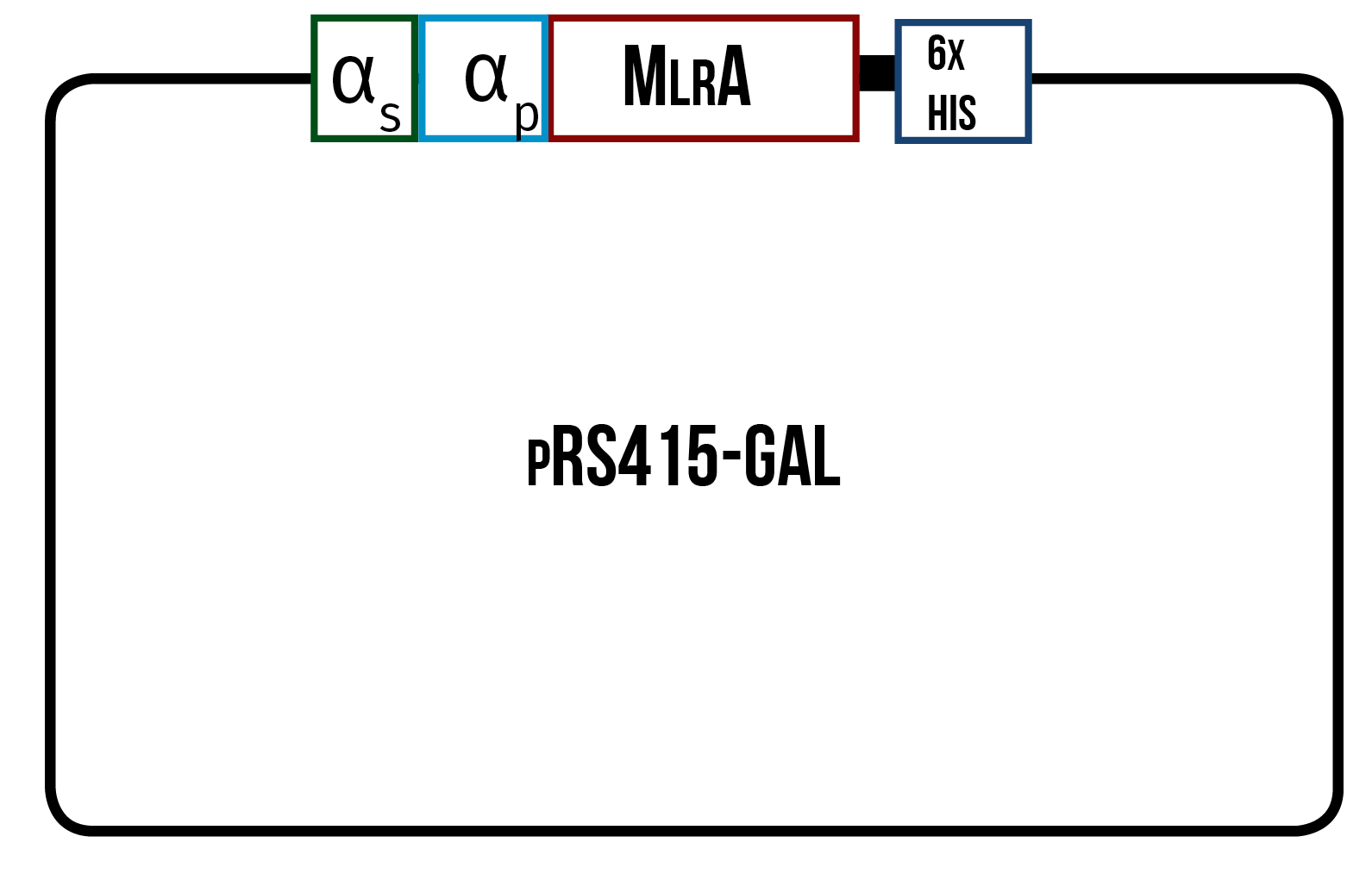
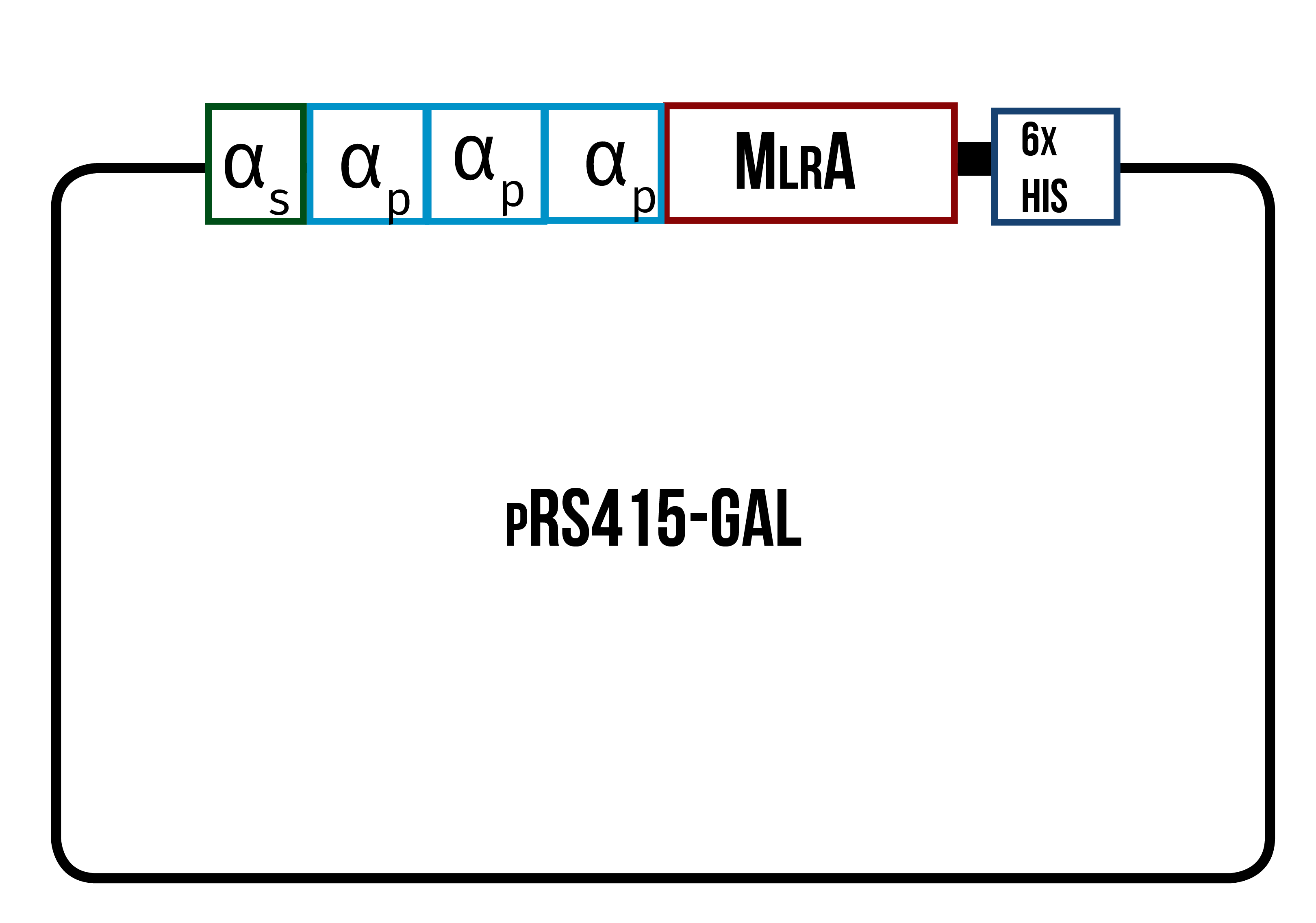
The MlrA sequence we decided to use is from Sphingomonas sp. ACM-3962 (GenBank accession number AF411068.2), as it had been previously used by Dziga et al. (2012) in the heterologous production of MlrA in E. coli . Since the original sequence is from a bacterial host, the sequence was codon optimized for yeast in hopes of achieving the highest possible yields in protein production. Codon optimization was done by GeneArt, from where we also ordered the constructs.
As it had already shown that MlrA can be produced in E. coli in an active form, we decided to produce that in addition to our yeast constructs as a positive control. With this positive control, we would be able to test our experimental setups with an enzyme we know should function correctly. This way, we would know if any potential problems when using the yeast constructs are due to the experimental setup or the actual enzyme. We based the design of our E. coli construct on what had been used by Dziga et al. (2012) in order to have maximal certainty that it would work. We designed our MlrA insert by fusing the MlrA sequence to a C-terminal His-tag (figure 15). The MlrA sequence was also codon optimized for E. coli , again by GeneArt.

In a previous iGEM project (“Ranger amongst Enemies”, Peking 2014), the MlrA enzyme was also produced heterologously in E. coli . They had created a BioBrick (BBa_1378001) of this MlrA sequence, but they had not specified the exact Sphingomonas sp. strain they had used. We believe that the reference sequence is the same as the one we chose to use ( Sphingomonas sp. ACM-3962), as of all the available MlrA sequences we compared, no other sequence corresponded to it as closely; the Peking 2014 biobrick differed from this sequence by only one amino acid. Because the difference of this single amino acid wasn’t explained, and no other sequence corresponded better to the Peking 2014 sequence, we assumed this single difference to be a mistake either in entering the BioBrick sequence, or due to a mutation that had happened during cloning. As a part of our gold medal criterion, we decided that we would improve this part (our improved part BBa_K1907002).
In the design of the degradation part of our project, we decided not to use the Peking BioBrick for our E. coli construct as it had the unexplained single amino acid difference to the closest available sequence. For our plasmid backbones, we decided to use ones we had available in our lab: to express MlrA in E. coli , we used pET28a, which is well suited for efficient IPTG-inducible protein expression in E. coli strain BL21(DE3). It is a similar plasmid to the pET21a used by Dziga et al. in their successful experiments, with only minor differences such as antibiotic resistance.
For yeast, as there were no available plasmid backbones for yeast in the BioBricks registry, we decided to use pRS415-GAL. This plasmid is well suited for protein expression inducible by galactose. Although we could have designed our insert to be cloned into the backbone using the XbaI restriction enzyme on the 5’ end, we reasoned that using biobrick-compatible restriction sites would potentially undermine the expression of our construct. Having the XbaI restriction site directly upstream of the protein coding sequence, as the use of the BioBricks suffix requires, would result in a T as the -3 base upstream of the start codon. It has been reported that in eukaryotic translation initiation, the -3 base is optimally an A or G (Kozak, 1996). This base is key in defining translation initiation, as changing it into a T or C increases sensitivity to differences in other bases upstream of the start codon. Cavener et al. (1991) on the other hand showed that yeast has a strong bias for A in this position. Nakagawa et al. (2008) concurred with this conclusion, in addition to pointing out a strong bias for G as the base following the start codon. Because our protein sequence didn’t contain this feature, we reasoned that having a suitable upstream sequence would be doubly important.
As fusing an additional, designed consensus sequence upstream of the protein coding sequence could have unforeseen consequences, we decided to clone our inserts into the plasmid using the SpeI and XhoI restriction sites (5’ and 3’ respectively), as these had been previously used in our lab to successfully express proteins in the same plasmid. Coincidentally, having a SpeI directly upstream of the start codon results in a favorable base (A) in the -3 position. Although this meant incompatibility with the BioBrick RFC 10 assembly standard, we decided it was better to be certain that any potential failure to express the protein wasn’t due to an unsuitable translation initiation site. Our plan was to test our constructs this way, but create BioBrick RFC 10 compatible parts for shipping to the registry. However, because of the issue of optimal eukaryotic translation initiation, we find the BioBrick assembly standard to be inconvenient for yeast BioBricks and protein expression in yeast.
About our final design
Cyanobacteria release their toxins only when they die and lyse. This poses a problem if one would add only microcystinase in water containing cyanobacteria and their toxins; the enzyme would only degrade the toxins that are floating freely in the water, but the toxins inside the intact bacteria would remain whole and potentially cause harm when the water is used. This is why the bacteria should also be lysed to ensure that all possible microcystin is available for the MlrA to be degraded. Based on the Peking 2014 project, hen egg lysozyme could be used to lyse the peptidoglycan cell walls of the cyanobacteria. Our idea is to use the same lysozyme from a commercially available source, at a low concentration so that it is safe for the user. In contrast to the Peking 2014 project, our enzyme capsule is designed for a much smaller volume of water and we expect it to be more productive. As the capsule is designed only for small volumes of water, the amount of lysozyme wouldn’t be harmful even if it ends up in the environment.
Our final product would thus be a capsule composed of microcystinase which we have expressed and purified, and lysozyme. Our product would be used only for relatively small volumes of water, i.e. it would not be used to e.g. purify a whole lake or make water drinkable, but only to detoxify a bucket of water used for recreational activities. From the degradation activity measurements, we would be able to calculate the amounts of MlrA needed to degrade a certain amount of MC-LR.
“ In order to find the conditions where the enzyme is produced best, we designed three different variants of the enzyme ”
References
Ahmad M, Hirz M, Pichler H, Schwab H., 2014. Protein expression in
Pichia pastoris
: recent achievements and perspectives for heterologous protein production. Appl Microbiol Biotechnol, 98(12), pp. 5301–5317.
Bitter, G. A., Chen, K. K., Banks, A. R., Lai, P.-H., 1984. Secretion of foreign proteins from
Saccharomyces cerevisiae
directed by a-factor gene fusions. Proc. Natl. Acad. Sci. USA Vol. 81, pp. 5330-5334.
Bourne DG, Riddles P, Jones GJ, Smith W, Blakely RL., 2001. Characterisation of a gene cluster involved in bacterial degradation of the cyanobacterial toxin microcystin LR. Environ Toxicol. 16(6), pp. 523-34.
Bourne DG, Jones GJ, Blakeley RL, Jones A, Negri AP, Riddles P., 1996. Enzymatic Pathway for the Bacterial Degradation of the Cyanobacterial Cyclic Peptide Toxin Microcystin LR. Appl. Environ. Microbiol. 62(11), pp. 4086-4094.
Campos, A., & Vasconcelos, V. 2010. Molecular Mechanisms of Microcystin Toxicity in Animal Cells. International Journal of Molecular Sciences, 11(1), pp. 268–287.
Cavener, D.R., Ray, S.C, Eukaryotic start and stop translation sites. Nucleic Acids Research, 1991, 19(12), pp. 3185-3192
Chorus I, Bartram J, eds., 1999. Toxic Cyanobacteria in Water: A Guide to Their Public Health Consequences, Monitoring, and Management. London: E & FN Spon.
Dziga D, Wladyka B, Zielińska G, Meriluoto J, Wasylewski M., 2012. Heterologous expression and characterisation of microcystinase. Toxicon, 59(5), pp. 578-586.
Estruch, F., 2000. Stress-controlled transcription factors, stress-induced genes and stress tolerance in budding yeast. FEMS microbiology reviews, 24(4), pp.469-486.
Faltermann, S., Prétôt, R., Pernthaler, J. and Fent, K., 2016. Comparative effects of nodularin and microcystin-LR in zebrafish: 1. Uptake by organic anion transporting polypeptide Oatp1d1 (Slco1d1). Aquatic Toxicology, 171, pp.69-76..
Farrugia, G., & Balzan, R. 2012. Oxidative Stress and Programmed Cell Death in Yeast. Frontiers in Oncology, 2, 64.
Güldener, U., Heck, S., Fiedler, T., Beinhauer, J. and Hegemann, J.H., 1996. A new efficient gene disruption cassette for repeated use in budding yeast. Nucleic acids research, 24(13), pp.2519-2524.
He, X.J. and Fassler, J.S., 2005. Identification of novel Yap1p and Skn7p binding sites involved in the oxidative stress response of
Saccharomyces cerevisiae
. Molecular microbiology, 58(5), pp.1454-1467.
Iwase, T., Tajima, A., Sugimoto, S., Okuda, K.I., Hironaka, I., Kamata, Y., Takada, K. and Mizunoe, Y., 2013. A simple assay for measuring catalase activity: a visual approach. Scientific reports, 3.
Kelley LA, Mezulis S, Yates CM, Wass MN, Sternberg MJE., 2015. The Phyre2 web portal for protein modeling, prediction and analysis. Nature Protocols 10, pp. 845–858.
Kozak, M. Point mutations define a sequence flanking the AUG initiator codon that modulates translation by eukaryotic ribosomes. 1996. Cell, 44(2), pp. 283-292
Nakagawa, S., Niimura, Y., Gojobori, T., Tanaka, H., Miura, K. Diversity of preferred nucleotide sequences around the translation initiation codon in eukaryote genomes. 2008. Nucleic Acids Research, 36(3), pp. 861-871
Plumer, Brad. "Toxic Algae Leaves Toledo without Drinking Water." Vox. -, 03 Aug. 2014. Accessed Web. 17 Oct. 2016.
Shaner, N.C., Steinbach, P.A. and Tsien, R.Y., 2005. A guide to choosing fluorescent proteins. Nature methods, 2(12), pp.905-909.
Valério, E., Vilares, A., Campos, A., Pereira, P. and Vasconcelos, V., 2014. Effects of microcystin-LR on
Saccharomyces cerevisiae
growth, oxidative stress and apoptosis. Toxicon, 90, pp.191-198.
Valério, E., Campos, A., Osório, H. and Vasconcelos, V., 2016. Proteomic and Real-Time PCR analyses of
Saccharomyces cerevisiae
VL3 exposed to microcystin-LR reveals a set of protein alterations transversal to several eukaryotic models. Toxicon, 112, pp.22-28.
Welgamage Don DON, A. C. D., 2012. An investigation into the biodegradation of peptide cyanotoxins (microcystins and nodularin) by novel gram-positive bacteria. Available from OpenAIR@RGU. [online]. Available from: http://openair.rgu.ac.uk
WHO, 2003. Cyanobacterial toxins: Microcystin-LR in drinking-water. Background document for preparation of WHO Guidelines for drinking-water quality. Geneva, World Health Organization (WHO/SDE/WSH/03.04/57).