Team:TU-Eindhoven/Modeling/PPI

In order to determine the properties of our designed protein pairs that cannot be directly measured, like the binding affinities/dissociation constants and cooperativity of our designed orthogonal pairs, we have developed a model based on mass-action and Michealis-Menten kinetics.
Our readouts will be done with our system in a quasi-steady state, so that is what our model will also be based on: It will be divided in 2 separate segments, the first will determine the quasi-steady state concentrations of all the proteins and protein-complexes in our system, the second will simulate the total activity based on the combined concentrations of the active complexes (both scaffold and non-scaffold mediated) in our system.
In order to find the quasi-steady state approximation of the system we must analyze the enter cascade of complex-formations starting from the base proteins/ligands (see figure 1). The steady state assumption allows us to determine the mass-action kinetics between the proteins with dissociation constants. Dissociation constants present in our system are between T14-3-3 and fusicoccin (KD,Fc)and between the T14-3-3-FC complex and the CT52s (KD,C1 and KD,C2), The Dissociation constants must be separated since they are not identical and thus do not have the same binding affinity to their respective T14-3-3 monomers, and the Dissociation constant of the non scaffold mediated active complex (KD,NoB). Besides Dissociation constants, a cooperativity parameter σ is introduced to simulate the stimulating (σ > 1) or inhibitory (σ < 1) effects of the first CT52 on the scaffold on the binding of the second CT52.
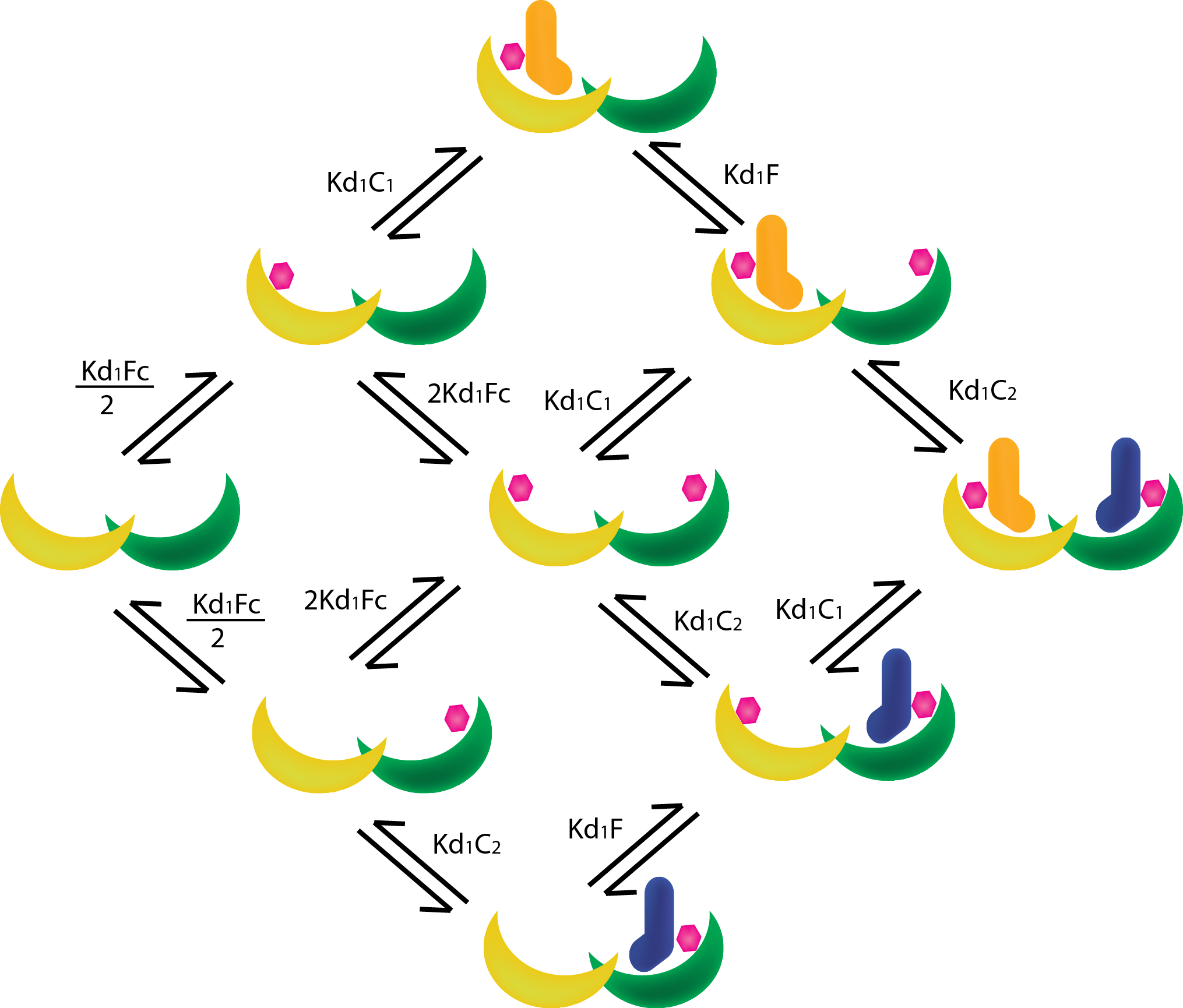
From figure 1 the mass-action equations can be derived. For the ease of notation the T14-3-3 scaffold will be represented as B, fusicoccin as F, and CT52 by C, followed by a number (1 or 2) if necessary. The following equations can be derived:

And non scaffold mediated:

Using the law of mass conservation we can setup equations for the total concentrations of the separate proteins and ligands in our system, which we can control during our assay:

By substituting equations 1-13 in equations 14-17, we get (ridiculously lengthy) equations describing the free concentrations of the system:

In order to solve equations 18 – 21 which contain multiple unknown variables, fixed point iteration is used using the initial conditions we use during our assays. Once equations 18-21 do not significantly change anymore we can assume the system is in a steady state and by using the results of the fixed point iteration we can calculate the concentrations of the complex species using equations 1-13, and finish the first segment of our model.
In order to determine the activity of the active complexes, we first state that concentration of enzyme [E] is the sum concentrations of scaffold (BFFCC) and non-scaffold (CC) mediated complexes. Next we use Michaelis-Menten kinetics to determine the initial concentration of the enzyme-substrate complex:

Using this initial enzyme-substrate concentration and the catalytic rate (kcat) of the active enzyme, the initial reaction rate can be calculated. To convert this reaction rate (which is calculated in μM/s) into the activity which is measured in the assay and then calibrated using a substrate specific calibration curve, the following formula is used:

With Mw the molecular weight of the fused C9-CT52 protein (the molecular weight of different variants of CT-52 are assumed to be the same), the resulting activity is expressed in U/mg.




