Difference between revisions of "Team:Toulouse France/Experiments"
| (55 intermediate revisions by 3 users not shown) | |||
| Line 2: | Line 2: | ||
{{Toulouse_France/menu}} | {{Toulouse_France/menu}} | ||
| + | <html> | ||
| + | |||
| + | <style> | ||
| + | .texteb { | ||
| + | color: black; | ||
| + | font-family: 'Open Sans'; | ||
| + | font-size: 15px; | ||
| + | margin: 20px 10px; | ||
| + | line-height: 24px; | ||
| + | text-align: justify; | ||
| + | } | ||
| + | .title1 { | ||
| + | color: #870721; | ||
| + | font-family: 'Open Sans'; | ||
| + | font-weight: 600; | ||
| + | font-size: 30px; | ||
| + | margin: 0 0 33px 0; | ||
| + | border: none; | ||
| + | text-align:center; | ||
| + | } | ||
| + | |||
| + | </style> | ||
| − | |||
<div class="page" style="background-image:url('https://static.igem.org/mediawiki/2016/4/4b/Toulouse_France_LascauxFresqueTaureaux.png');"> | <div class="page" style="background-image:url('https://static.igem.org/mediawiki/2016/4/4b/Toulouse_France_LascauxFresqueTaureaux.png');"> | ||
| Line 14: | Line 35: | ||
<div id="pageintro" class="hoc clear" style="padding:300px 0px;"> | <div id="pageintro" class="hoc clear" style="padding:300px 0px;"> | ||
| − | <p class="sec_title" style="background-color:rgba(1,1,1,0.5);"> | + | <p class="sec_title" style="background-color:rgba(1,1,1,0.5);">Results</p> |
</div> | </div> | ||
| Line 21: | Line 42: | ||
</div> | </div> | ||
<!-- ################################################################################################ --> | <!-- ################################################################################################ --> | ||
| + | |||
| − | < | + | |
| + | <div class="column full_size" style="background-color:#F5F5F5;"> | ||
| + | <center><hr style="width:70%; margin:10px 0px; color:black; background-color:black; height:1px; align:center;" /> </center> | ||
| + | </div> | ||
| + | |||
| + | <div class="column full_size" id="predation" style="background-color:#F5F5F5; text-align:justify; padding:10px 10%;"> | ||
| − | + | <u><p class="title1" id="select1">Predation</p></u> | |
| + | |||
| + | |||
| + | <p class="texteb"> | ||
| + | |||
| + | Our aim is to reinforce the natural predation capacity of <i>B. subtilis</i> and to ensure it is expressed independantly of the conditions. We | ||
| − | + | first assessed that our wild type <i>Bacillus</i> chassis is not able of predation, then we built the operons | |
| − | + | allowing boosting the predation property. | |
| + | <br><br> | ||
| + | |||
| + | <u><b>Preliminary tests:</b></u> | ||
| + | <br><br>We tried different testing approaches to evaluate the predatory response of <i>B. subtilis</i> and eventually elaborate a protocol to do the preliminary tests. We tested the predation of <i>B. subtilis</i> Wild Type (strain 168) against <i>Pseudomonas fluorescens</i> (strain SBW25), a deleterious strain present in the cave. Briefly, the protocol consists in growing both strains in rich medium, mixing them in PBS and monitoring their growth (figure 1). | ||
| + | </p> | ||
| + | |||
| + | <p> | ||
| + | <!-- ###### FIGURE ##### --> | ||
| + | <center><img src="https://static.igem.org/mediawiki/2016/2/22/Toulouse_France_results1.png" style="width:60%; margin:20px 20px;"></img> | ||
| + | <b> | ||
| + | <br>Figure 1: <i>Bacillus subtilis</i> WT 168 does not feed on <i>Pseudomonas fluorescens</i> SBW25. Both strains were grown overnight in rich medium and then mixed in PBS. The growths of the strains were then monitored during 8 hours by plate numeration. The graph represents the ratio between <i>B. subtilis</i> in PBS in presence of <i>P. fluorescens</i> versus <i>B. subtilis</i> alone in PBS (data normalized to time 1H). | ||
| + | </b></center> | ||
| + | |||
| + | <br><br> | ||
| + | </p> | ||
| + | |||
| + | |||
| + | <p class="texteb"> | ||
| + | We observed no growth benefit when mixing <i>B. subtilis</i> and <i>P. fluorescens</i> compared to <i>B. subtilis</i> alone. We conclude that <i>B. subtilis</i> WT predation program is not the strain priority when facing starvation. Other surviving program as competence or sporulation are likely favoured by <i>B. subtilis</i> in such condition. This reinforces the need to prevent these programs by using a <i>spo0A</i> mutant and to promote the predation by overexpressing either the SKF or SDP operons. | ||
| + | <br><br> | ||
| + | |||
| + | <u><b>SKF</b></u> | ||
| + | <br><br> | ||
| + | This predation operon is composed of seven genes for a total of more than 6 kb. To get rid of restriction sites that could interfere with the cloning steps, we ordered the optimized sequences from IDT as four gblocks. From there, our strategy was to do Gibson cloning to obtain the full operon in pSB1C3 in <i>E. coli</i> and then to transfer it in <i>B. subtilis</i>. However, we did not manage to obtain the whole assembly (figure 2), neither partial ones, in spite of about 20 attempts… | ||
| + | </p> | ||
| − | + | <p> | |
| + | <!-- ###### FIGURE ##### --> | ||
| + | <center><img src="https://static.igem.org/mediawiki/2016/2/28/Toulouse_France_results2.png" style="width:80%; margin:20px 20px;"> | ||
| + | <b> | ||
| + | <br>Figure 2 : Layout of SKF expected biobrick. | ||
| + | </b></center> | ||
| + | <br><br> | ||
| + | </p> | ||
| + | |||
| + | <p class="texteb"> | ||
| + | <u><b>SDP</b></u> | ||
| + | <br><br> | ||
| + | The SDP operon is smaller than the SKF one and it was possible to obtain the optimized sequences as two gblocks. Here again, we were unfortunate and did not get the expected clones in <i>E. coli</i> (figure 3). | ||
| + | </p> | ||
| + | |||
| + | <p> | ||
| + | <!-- ###### FIGURE ##### --> | ||
| + | <center><img src="https://static.igem.org/mediawiki/2016/e/e0/Toulouse_France_results3.png" style="width:70%; margin:20px 20px;"> | ||
| + | <b> | ||
| + | <br>Figure 3 : Layout of SDP expected biobrick. | ||
| + | </b></center><br><br> | ||
| + | To perform trouble shooting, we tried an assembly test with just the two gblocks and deposited the product on gel. We observed that the reaction seems to be effective with the presence of a new band corresponding to the combined size of the two gblocks (figure 4). | ||
| − | + | <!-- ###### FIGURE ##### --> | |
| − | + | <center><img src="https://static.igem.org/mediawiki/2016/9/96/Toulouse_France_results4.png" style="width:30%; margin:20px 20px;"> | |
| + | <b> | ||
| + | <br>Figure 4: Gibson assemby of the two SDP Gblocks. | ||
| + | </b></center><br><br> | ||
| + | </p> | ||
| + | |||
| + | <p class="texteb"> | ||
| + | |||
| + | <u><b>Conclusions and perspectives</b></u> | ||
| + | <br><br> | ||
| + | It seems our Gibson step is fine since we managed to obtain the SDP assembly, but we could not get <i>E. coli</i> transformants when performing the whole experience. The predation system is based on the production of toxins by <i>B. subtilis</i>, and these toxins were reported to be harmful to <i>E. coli</i> (Nandy et al., 2007, FEBS Letters. 581: 151–56). An explanation to our problems could be that SDP and SKF cloning in <i>E. coli</i> results in the bacterium death. We had thought about this problem, but we had believed the expression driven by the pVeg <i>Bacillus</i> promoter to be insufficient for such effect. Perspectives could be to use a tightly regulated promoter to prevent expression during the cloning step in <i>E. coli</i>, or to try a direct transformation of highly competent <i>Bacillus</i> strain. | ||
| + | </p> | ||
| − | + | </div> | |
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| + | <div class="column full_size" style="background-color:#F5F5F5;"> | ||
| + | <center><hr style="width:70%; margin:10px 0px; color:black; background-color:black; height:1px; align:center;" /> </center> | ||
| + | </div> | ||
| + | |||
| + | <div class="column full_size" id="antifongique" style="background-color:#F5F5F5; text-align:justify; padding: 10px 10%"> | ||
| − | + | ||
| − | + | <p class="title1" id="select1">Antifungals<p> | |
| + | |||
| + | <p class="texteb"> | ||
| + | Here, we aimed to produce a cocktail of five antifungal peptides whose production in <i>Bacillus subtilis</i> will be triggered by presence of fungi. | ||
| + | <br> | ||
| + | <br> | ||
| + | <u><b style="font-size:16px;">Operon constructions:</b></u> | ||
| + | </b><br><br> | ||
| + | The whole antifungal operon was too big to be synthesized by IDT as one gblock. We therefore decided to divide it in two operons (figure 5), each of them with a promoter to be functional, with the possibility to eventually combine them. The sequence were optimized for the <i>Bacillus</i> codon usage and to remove inadequate restriction sites. Sub-cloning of the first operon (containing cut version of the Metchnikowin and D4E1) on the pSB1C3 backbone was rapidly performed, leading to the new composite part <a href="http://parts.igem.org/Part:BBa_K1937007">BBa_K1937007 (pSB1C3-AF_A)</a>. However, we did not manage to obtain the second operon in the pSB1C3 (encoding Dermaseptin B1, GAFP-1 and entire Metchnikowin antifungal peptides). We tried to directly sub-clone the gblock in the pSB1C3-AF_A but without success. We hypothesize that one of the peptide could be toxic for <i>E. coli</i>. This will have to be verified by sub-cloning the 3 peptides alone. The AF_A operon was subsequently cloned in the pSB<sub>BS</sub>0K-Mini plasmid to create biobrick <a href="http://parts.igem.org/Part:BBa_K1937008">BBA_K1937008</a>. | ||
| + | </p> | ||
| + | |||
| + | <p> | ||
| + | <!-- ###### FIGURE ##### --> | ||
| + | <center><img src="https://static.igem.org/mediawiki/2016/d/dc/Toulouse_France_results5.png" style="width:70%; margin:20px 20px;"> | ||
| + | <b> | ||
| + | <br>Figure 5: Layout of antifungal operons and their assembly. | ||
| + | </b></center><br><br> | ||
| + | </p> | ||
| + | |||
| + | <p class="texteb"> | ||
| + | In order to express specifically the antifungal peptides in close vicinity to fungi, we choose the two N-acetyl-glucosamine (NAG) inducible promotors pNagA and pNagP. The constructions with the RFP reporter gene were ordered from IDT and successfully sub-cloned in the pSB1C3 (new parts <a href="http://parts.igem.org/Part:BBa_K1937003">BBa_K1937003</a> and <a href="http://parts.igem.org/Part:BBa_K1937005">BBa_K1937005</a> ; figure 6). They were subsequently cloned in the pSB<sub>BS</sub>0K-Mini plasmid to create biobricks <a href="http://parts.igem.org/Part:BBa_K1937004">BBA_K1937004</a> and <a href="http://parts.igem.org/Part:BBa_K1937006">BBa_K1937006</a>. | ||
| + | </p> | ||
| − | + | <p> | |
| − | + | <!-- ###### FIGURE ##### --> | |
| − | + | <center><img src="https://static.igem.org/mediawiki/2016/4/41/Toulouse_France_results6.png" style="width:50%; margin:20px 20px;"> | |
| − | + | <b> | |
| − | + | <br>Figure 6: Layout of the pNag-RFP constructions. | |
| − | + | </b></center><br><br> | |
| − | + | </p> | |
| − | + | ||
| − | + | <p class="texteb"> | |
| − | + | <u><b>pNag validation</b></u> | |
| − | + | <br><br> | |
| − | + | We tested the expression and specificity of the RFP driven by pNagA and pNagP when growing in presence of glucose or NAG (figure 7). We observed a late and rather specific RFP expression on NAG. The late expression could mean that the formulation of our minimal medium is not optimal. The fact that the pNagA-RFP and pNagP-RFP strains seem able to slightly express the RFP on glucose (figure 7B, left panel close-up), albeit on weaker extend that on NAG (figure 7B, right panel close-up), could be due to the alleviating of the catabolic repression. | |
| − | + | <br><br>In conclusion, pNagA and pNagP appear as able to promote expression in response to NAG, even if the growth conditions could be improved to get higher and more homogeneous expression levels. | |
| − | + | ||
| − | + | ||
| − | + | </p> | |
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| + | <p> | ||
| + | <!-- ###### FIGURE ##### --> | ||
| + | <center><img src="https://static.igem.org/mediawiki/2016/4/4b/Toulouse_France_results7.png" style="width:40%; margin:20px 20px;"> | ||
| + | <b> | ||
| + | <br>Figure 7: NAG-driven expression of RFP. <i>B. subtilis</i> strains transformed with pSB<sub>BS</sub>0K-Mini (Control), pSB<sub>BS</sub>0K-Mini-NagA or pSB<sub>BS</sub>0K-Mini-NagP were spread on minimal medium with either glucose or NAG as carbon source. Red spots appeared only with pNagA or pNagP on NAG (close-ups on part 7B). | ||
| + | </b></center><br><br> | ||
| + | </p> | ||
| + | <p class="texteb"> | ||
| + | <u><b>Antifungal validation</b></u> | ||
| + | <br><br> | ||
| + | We found out that the best culture conditions for the fungi that permits a slight growth of <i>Bacillus</i> were with ¼ PDA and 2% glucose. We tested different fungi (<i>Aspergillus niger, Talaromyces funiculosus</i> and <i>Chaetomium globosum</i>) but we eventually focussed on <i>Talaromyces funiculosus</i> that seems easier to manipulate to us. | ||
| − | + | <br><br>Our test consisted in adding, on fungi inoculated plates, paper patches soaked with either copper sulfate (positive control), LB medium (negative control), a suspension of <i>Bacillus subtilis</i> WT or <i>Bacillus subtilis</i> expressing the antifungal AF_A operon (figure 8). We observed that with our construction, a slight inhibition halo appeared around the patch. This effect is visible even after 8 days and was reproducible. These observations allow us to conclude that AF_A is functional. | |
| − | + | </p> | |
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | <p> | |
| − | + | <!-- ###### FIGURE ##### --> | |
| − | + | <center><img src="https://static.igem.org/mediawiki/2016/9/99/Toulouse_France_results8.png" style="width:50%; margin:20px 20px;"> | |
| − | + | <b> | |
| − | + | <br>Figure 8: Antifungal tests (legend in the text). | |
| − | + | </b></center><br><br> | |
| − | + | </p> | |
| − | + | ||
| − | + | <p class="texteb"> | |
| − | + | <u><b>Test on the rock</b></u> | |
| − | + | <br><br> | |
| − | + | As our therapeutic bacterium was supposed to treat fungi growing on the walls of a cave, we needed to test its activity in conditions that would mimic the cave’s environment. The experimental model we thought about was to test our modified bacteria on fungi artificially grown on rocks. The first step was therefore to be able to grow fungi on rocks. | |
| − | + | <br>In order to do so, we have deposed on the surface of the rocks growth media with various nutriment compositions (see Table 1). The red color of the spots was due to ochre, whose purpose was to mimic the frescoes of the cave (See Figure below). The spots 1 to 3 contain various concentrations of glucose, the spots 4 to 6 various concentrations of tryptone and yeast extracts, whereas the spots 7-8 various concentrations of glucose, tryptone and yeast extract. | |
| − | + | <br>The fungi were then inoculated on each spot whereas our therapeutic agent only on Spot 8. | |
| − | + | </p> | |
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | <p> | |
| − | + | <br><br> | |
| − | + | <center><img src="https://static.igem.org/mediawiki/2016/8/8b/Toulouse_France_TableauTestpierre2.jpg" style="width:50%; margin:20px 20px;"></center> | |
| − | + | <br><br> | |
| − | + | </p> | |
| − | + | ||
| − | + | <p class="texteb"> | |
| − | + | <u><b>Results</b></u> | |
| − | + | <br><br> | |
| − | + | </p> | |
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | <p> | |
| − | + | <center><img src="https://static.igem.org/mediawiki/2016/e/e5/Toulouse_France_testpierreT0.jpg" style="width:40%;"> | |
| − | + | <img src="https://static.igem.org/mediawiki/2016/6/6c/Toulouse_France_testpierreT3.jpg" style="width:30%;"> | |
| − | + | <b> | |
| − | + | <br>Figure 9: Test on the rock inoculated with <i>Talaromyces funiculosum</i> at T=0 (on the left) and T=3 weeks post infection (on the right). | |
| − | + | </b></center> | |
| − | + | </p> | |
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | <p class="texteb"> | |
| − | + | <br><br> | |
| − | + | After 3 weeks, the growth of fungi was clearly visible on spots 1 to 6, with the most efficient growth on the spot 3 which had the following medium composition: | |
| − | + | <ul> | |
| − | + | <li>1g Glucose </li> | |
| − | + | <li>1g Yeast Extract </li> | |
| − | + | <li>1g Tryptone </li> | |
| − | + | </ul> | |
| − | + | ||
| − | + | ||
| − | + | <br><br> | |
| − | + | Interestingly, no growth of the fungus was observed on spots 7 and 8. As the later one contained our therapeutic agent, this observation suggested that our modified bacterium might be active against the fungi. However, the absence of growth on Spot 7 would argue against this conclusion. It is therefore clear that this result needs to be confirmed by repeating the experiment several times. However, the ability to get the fungi grown on rock was already a good result, indicating that we might have a good model to test our bacteria. | |
| − | + | </p> | |
| − | + | ||
| − | + | ||
| − | + | <p class="texteb"> | |
| − | + | <u><b>Conclusions and perspectives</b></u> | |
| − | + | <br><br>Here, we showed that our pNagA and NagP parts are able to control gene expression in response to NAG and that the first part of our antifungal operon is functional. In both cases, the properties will have to be optimized, through a higher and more homogeneous expression from the NAG-driven promoters and through the completion of the antifungal operons to produce more than two antifungal peptides. | |
| − | + | <br><br>We were able to set up a model which mimics the cave's environment. Thanks to it, we got encouraging results showing that the therapeutic agent might be functional. | |
| − | + | <br><br> | |
| − | + | </p> | |
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | </div> | |
| − | + | ||
| − | + | <div class="column full_size" style="background-color:#F5F5F5;"> | |
| − | + | <center><hr style="width:70%; margin:10px 0px; color:black; background-color:black; height:1px; align:center;" /> </center> | |
| − | + | ||
| − | + | </div> | |
| − | + | ||
| − | + | <div class="column full_size" id="confinement" style="background-color:#F5F5F5; text-align:justify; padding: 0px 10%"> | |
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | <p class="title1" id="select1">Containement<p> | |
| − | + | <p class="texteb"> | |
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | Here, we fashioned a genetic system to prevent horizontal transfer of our synthetic constructions. | |
| + | <br><br> | ||
| + | <u><b>Toxin/antitoxin systems constructions</b></u> | ||
| + | <br><br> | ||
| + | The constructions were ordered as gblocks from IDT. The Epsilon/MazF construction was rapidly sub-cloned in the pSB1C3 backbone (new composite part <a href="http://parts.igem.org/Part:BBa_K1937009">BBa_K1937009</a>), and then in the pSB<sub>BS</sub>0K-Mini plasmid to create biobricks <a href="http://parts.igem.org/Part:BBa_K1937010">BBa_K1937010</a> (figure 10). However, we never managed to get the MazE/Zeta construction in the pSB1C3 backbone. Again, we can only speculate about the toxicity of the toxin. | ||
| + | </p> | ||
| + | |||
| + | <p> | ||
| + | <!-- ###### FIGURE ##### --> | ||
| + | <center><img src="https://static.igem.org/mediawiki/2016/e/e7/Toulouse_France_result.png" style="width:50%; margin:20px 20px;"> | ||
| + | <b> | ||
| + | <br>Figure 10: Layout of the toxin/antitoxin operons. | ||
| + | </b></center><br><br> | ||
| + | </p> | ||
| + | |||
| + | <p class="texteb"> | ||
| + | <u><b>Theophylline validation</b></u> | ||
| + | <br><br> | ||
| + | To validate the theophylline riboswitch, we inferred that we should obtain clones of <i>Bacillus subtilis</i> transformed with the pSB<sub>BS</sub>0K-Mini –Epsilon/MazF only in presence of theophylline: the molecule should prevent the expression of the MazF toxin that is lethal since the antitoxin MazE is not present. Unfortunately, we did not get any clone, neither without nor with theophylline (figure 11). | ||
| + | </p> | ||
| + | |||
| + | <p> | ||
| + | <!-- ###### FIGURE ##### --> | ||
| + | <center><img src="https://static.igem.org/mediawiki/2016/5/5b/Toulouse_France_results10.png" style="width:40%; margin:20px 20px;"> | ||
| + | <b> | ||
| + | <br>Figure 11: Result of the <i>Bacillus subtilis</i> transformation with pSB<sub>BS</sub>0K-Mini –Epsilon/MazF (we know this is not the most illustrative figure ever!). | ||
| + | </b></center><br><br> | ||
| + | </p> | ||
| + | |||
| + | <p class="texteb"> | ||
| + | <u><b>Conclusions and perspectives</b></u> | ||
| + | <br><br> | ||
| + | At this step, we can only hypothesize that our system is leaking sufficient expression of the toxins for them to be lethal, either in <i>E. coli</i> or in <i>B. subtilis</i>. Further assays using inducible promoters will be necessary to set up the system without enduring these toxicity problems. | ||
| + | </p> | ||
| + | |||
| + | </div> | ||
| − | + | <div class="column full_size" style="background-color:#F5F5F5;"> | |
| − | + | <center><hr style="width:70%; margin:10px 0px; color:black; background-color:black; height:1px; align:center;" /> </center> | |
| − | + | </div> | |
| + | |||
| + | <div class="column full_size" style="background-color:#F5F5F5;"> | ||
| + | |||
| + | <center> | ||
| + | <a class="button-home" href="https://2016.igem.org/Team:Toulouse_France/Description" style="border: 1px solid #282828;-webkit-border-radius: 5px;-moz-border-radius: 5px;border-radius: 5px; | ||
| + | padding: 15px 15px; color: black; text-decoration: none; font-size: 18px; background: none; display: block; width: 250px; background-color:#3CB371">BACK: Backbones description</a> | ||
| + | <br><br> | ||
| + | <a class="button-home" href="https://2016.igem.org/Team:Toulouse_France/Model" style="border: 1px solid #282828;-webkit-border-radius: 5px;-moz-border-radius: 5px;border-radius: 5px; | ||
| + | padding: 15px 15px; color: black; text-decoration: none; font-size: 18px; background: none; display: block; width: 250px; background-color:#FF6347">NEXT: Modeling</a> | ||
| + | </center> | ||
| + | |||
| + | </div> | ||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
</html> | </html> | ||
| − | |||
{{Toulouse_France/Sponsors}} | {{Toulouse_France/Sponsors}} | ||
{{Toulouse_France/Footer}} | {{Toulouse_France/Footer}} | ||
Latest revision as of 02:39, 20 October 2016
Follow us @


![]()
Results
Predation
Our aim is to reinforce the natural predation capacity of B. subtilis and to ensure it is expressed independantly of the conditions. We
first assessed that our wild type Bacillus chassis is not able of predation, then we built the operons
allowing boosting the predation property.
Preliminary tests:
We tried different testing approaches to evaluate the predatory response of B. subtilis and eventually elaborate a protocol to do the preliminary tests. We tested the predation of B. subtilis Wild Type (strain 168) against Pseudomonas fluorescens (strain SBW25), a deleterious strain present in the cave. Briefly, the protocol consists in growing both strains in rich medium, mixing them in PBS and monitoring their growth (figure 1).
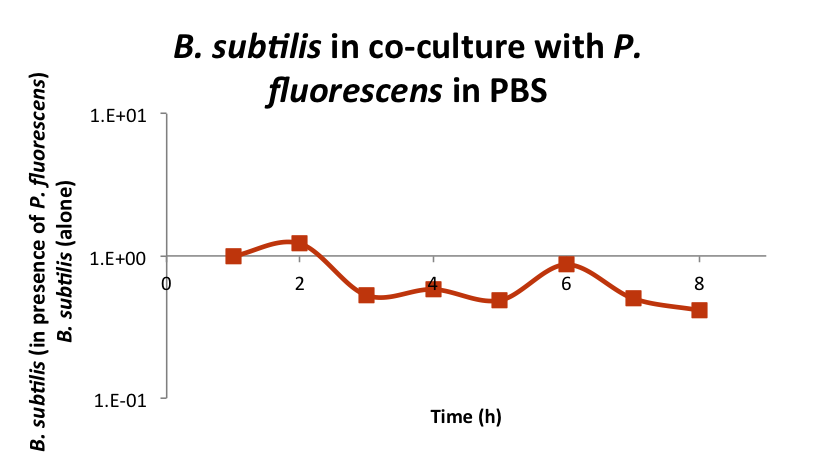
Figure 1: Bacillus subtilis WT 168 does not feed on Pseudomonas fluorescens SBW25. Both strains were grown overnight in rich medium and then mixed in PBS. The growths of the strains were then monitored during 8 hours by plate numeration. The graph represents the ratio between B. subtilis in PBS in presence of P. fluorescens versus B. subtilis alone in PBS (data normalized to time 1H).
We observed no growth benefit when mixing B. subtilis and P. fluorescens compared to B. subtilis alone. We conclude that B. subtilis WT predation program is not the strain priority when facing starvation. Other surviving program as competence or sporulation are likely favoured by B. subtilis in such condition. This reinforces the need to prevent these programs by using a spo0A mutant and to promote the predation by overexpressing either the SKF or SDP operons.
SKF
This predation operon is composed of seven genes for a total of more than 6 kb. To get rid of restriction sites that could interfere with the cloning steps, we ordered the optimized sequences from IDT as four gblocks. From there, our strategy was to do Gibson cloning to obtain the full operon in pSB1C3 in E. coli and then to transfer it in B. subtilis. However, we did not manage to obtain the whole assembly (figure 2), neither partial ones, in spite of about 20 attempts…

Figure 2 : Layout of SKF expected biobrick.
SDP
The SDP operon is smaller than the SKF one and it was possible to obtain the optimized sequences as two gblocks. Here again, we were unfortunate and did not get the expected clones in E. coli (figure 3).

Figure 3 : Layout of SDP expected biobrick.
To perform trouble shooting, we tried an assembly test with just the two gblocks and deposited the product on gel. We observed that the reaction seems to be effective with the presence of a new band corresponding to the combined size of the two gblocks (figure 4).

Figure 4: Gibson assemby of the two SDP Gblocks.
Conclusions and perspectives
It seems our Gibson step is fine since we managed to obtain the SDP assembly, but we could not get E. coli transformants when performing the whole experience. The predation system is based on the production of toxins by B. subtilis, and these toxins were reported to be harmful to E. coli (Nandy et al., 2007, FEBS Letters. 581: 151–56). An explanation to our problems could be that SDP and SKF cloning in E. coli results in the bacterium death. We had thought about this problem, but we had believed the expression driven by the pVeg Bacillus promoter to be insufficient for such effect. Perspectives could be to use a tightly regulated promoter to prevent expression during the cloning step in E. coli, or to try a direct transformation of highly competent Bacillus strain.
Antifungals
Here, we aimed to produce a cocktail of five antifungal peptides whose production in Bacillus subtilis will be triggered by presence of fungi.
Operon constructions:
The whole antifungal operon was too big to be synthesized by IDT as one gblock. We therefore decided to divide it in two operons (figure 5), each of them with a promoter to be functional, with the possibility to eventually combine them. The sequence were optimized for the Bacillus codon usage and to remove inadequate restriction sites. Sub-cloning of the first operon (containing cut version of the Metchnikowin and D4E1) on the pSB1C3 backbone was rapidly performed, leading to the new composite part BBa_K1937007 (pSB1C3-AF_A). However, we did not manage to obtain the second operon in the pSB1C3 (encoding Dermaseptin B1, GAFP-1 and entire Metchnikowin antifungal peptides). We tried to directly sub-clone the gblock in the pSB1C3-AF_A but without success. We hypothesize that one of the peptide could be toxic for E. coli. This will have to be verified by sub-cloning the 3 peptides alone. The AF_A operon was subsequently cloned in the pSBBS0K-Mini plasmid to create biobrick BBA_K1937008.

Figure 5: Layout of antifungal operons and their assembly.
In order to express specifically the antifungal peptides in close vicinity to fungi, we choose the two N-acetyl-glucosamine (NAG) inducible promotors pNagA and pNagP. The constructions with the RFP reporter gene were ordered from IDT and successfully sub-cloned in the pSB1C3 (new parts BBa_K1937003 and BBa_K1937005 ; figure 6). They were subsequently cloned in the pSBBS0K-Mini plasmid to create biobricks BBA_K1937004 and BBa_K1937006.

Figure 6: Layout of the pNag-RFP constructions.
pNag validation
We tested the expression and specificity of the RFP driven by pNagA and pNagP when growing in presence of glucose or NAG (figure 7). We observed a late and rather specific RFP expression on NAG. The late expression could mean that the formulation of our minimal medium is not optimal. The fact that the pNagA-RFP and pNagP-RFP strains seem able to slightly express the RFP on glucose (figure 7B, left panel close-up), albeit on weaker extend that on NAG (figure 7B, right panel close-up), could be due to the alleviating of the catabolic repression.
In conclusion, pNagA and pNagP appear as able to promote expression in response to NAG, even if the growth conditions could be improved to get higher and more homogeneous expression levels.

Figure 7: NAG-driven expression of RFP. B. subtilis strains transformed with pSBBS0K-Mini (Control), pSBBS0K-Mini-NagA or pSBBS0K-Mini-NagP were spread on minimal medium with either glucose or NAG as carbon source. Red spots appeared only with pNagA or pNagP on NAG (close-ups on part 7B).
Antifungal validation
We found out that the best culture conditions for the fungi that permits a slight growth of Bacillus were with ¼ PDA and 2% glucose. We tested different fungi (Aspergillus niger, Talaromyces funiculosus and Chaetomium globosum) but we eventually focussed on Talaromyces funiculosus that seems easier to manipulate to us.
Our test consisted in adding, on fungi inoculated plates, paper patches soaked with either copper sulfate (positive control), LB medium (negative control), a suspension of Bacillus subtilis WT or Bacillus subtilis expressing the antifungal AF_A operon (figure 8). We observed that with our construction, a slight inhibition halo appeared around the patch. This effect is visible even after 8 days and was reproducible. These observations allow us to conclude that AF_A is functional.

Figure 8: Antifungal tests (legend in the text).
Test on the rock
As our therapeutic bacterium was supposed to treat fungi growing on the walls of a cave, we needed to test its activity in conditions that would mimic the cave’s environment. The experimental model we thought about was to test our modified bacteria on fungi artificially grown on rocks. The first step was therefore to be able to grow fungi on rocks.
In order to do so, we have deposed on the surface of the rocks growth media with various nutriment compositions (see Table 1). The red color of the spots was due to ochre, whose purpose was to mimic the frescoes of the cave (See Figure below). The spots 1 to 3 contain various concentrations of glucose, the spots 4 to 6 various concentrations of tryptone and yeast extracts, whereas the spots 7-8 various concentrations of glucose, tryptone and yeast extract.
The fungi were then inoculated on each spot whereas our therapeutic agent only on Spot 8.

Results


Figure 9: Test on the rock inoculated with Talaromyces funiculosum at T=0 (on the left) and T=3 weeks post infection (on the right).
After 3 weeks, the growth of fungi was clearly visible on spots 1 to 6, with the most efficient growth on the spot 3 which had the following medium composition:
- 1g Glucose
- 1g Yeast Extract
- 1g Tryptone
Interestingly, no growth of the fungus was observed on spots 7 and 8. As the later one contained our therapeutic agent, this observation suggested that our modified bacterium might be active against the fungi. However, the absence of growth on Spot 7 would argue against this conclusion. It is therefore clear that this result needs to be confirmed by repeating the experiment several times. However, the ability to get the fungi grown on rock was already a good result, indicating that we might have a good model to test our bacteria.
Conclusions and perspectives
Here, we showed that our pNagA and NagP parts are able to control gene expression in response to NAG and that the first part of our antifungal operon is functional. In both cases, the properties will have to be optimized, through a higher and more homogeneous expression from the NAG-driven promoters and through the completion of the antifungal operons to produce more than two antifungal peptides.
We were able to set up a model which mimics the cave's environment. Thanks to it, we got encouraging results showing that the therapeutic agent might be functional.
Containement
Here, we fashioned a genetic system to prevent horizontal transfer of our synthetic constructions.
Toxin/antitoxin systems constructions
The constructions were ordered as gblocks from IDT. The Epsilon/MazF construction was rapidly sub-cloned in the pSB1C3 backbone (new composite part BBa_K1937009), and then in the pSBBS0K-Mini plasmid to create biobricks BBa_K1937010 (figure 10). However, we never managed to get the MazE/Zeta construction in the pSB1C3 backbone. Again, we can only speculate about the toxicity of the toxin.

Figure 10: Layout of the toxin/antitoxin operons.
Theophylline validation
To validate the theophylline riboswitch, we inferred that we should obtain clones of Bacillus subtilis transformed with the pSBBS0K-Mini –Epsilon/MazF only in presence of theophylline: the molecule should prevent the expression of the MazF toxin that is lethal since the antitoxin MazE is not present. Unfortunately, we did not get any clone, neither without nor with theophylline (figure 11).

Figure 11: Result of the Bacillus subtilis transformation with pSBBS0K-Mini –Epsilon/MazF (we know this is not the most illustrative figure ever!).
Conclusions and perspectives
At this step, we can only hypothesize that our system is leaking sufficient expression of the toxins for them to be lethal, either in E. coli or in B. subtilis. Further assays using inducible promoters will be necessary to set up the system without enduring these toxicity problems.





Website by Team iGEM Toulouse 2016