Team:Newcastle/Notebook/Lab/ODE-Modelling
ODE Modelling
As part of our work, we performed many different types of modelling, from rule-based modelling of our genetic parts to physics analysis of our microfluidics components. On this page, we discuss the ordinary differential equation (ODE) modelling of our components for use in a web-based ‘simulator’. We hope that the ‘simulator’ will demonstrate how our genetic constructs could fit into an electronic circuit.
Simulator Core
The core of our simulator features a set of ODEs that describe an abstract version of the transcriptional (Equation 1) and translational (Equation 2) processes in a cell.
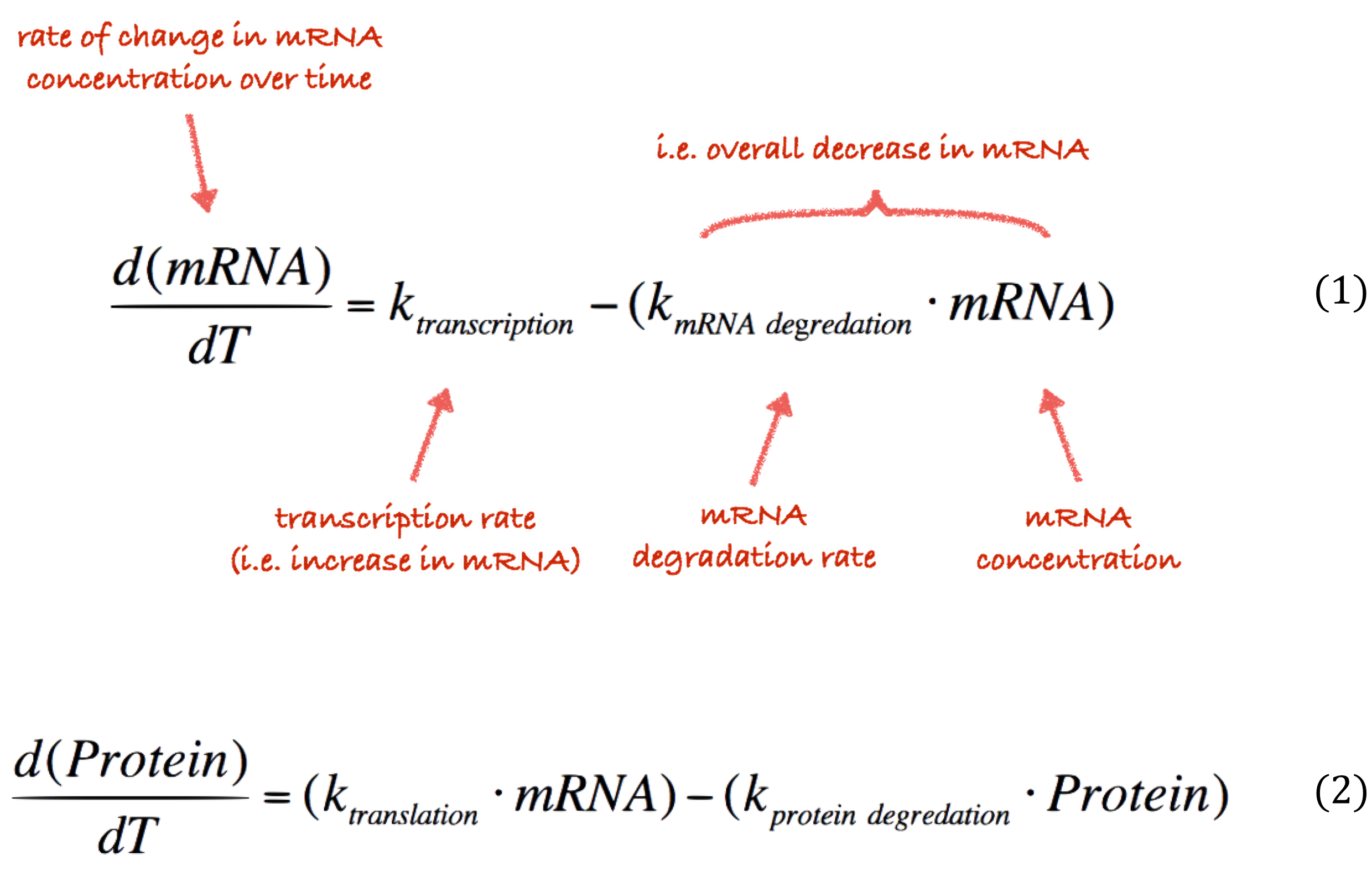
The rate at which these processes occur is controlled by a number of paramters, and . These constants describe the rate of transcription rate, degradation of mRNA, translation rate and protein degradation rate respectively. In our modelling we focus only on variation in the and parameters; we can fluctuate these values various promoter strengths and protein degradation tags. The other parameters are unrelated to any of the elements of our system that we control. Consequently, we have assigned these fixed values in our simulator.
Since our code is freely available, a modeller with different simulation requirements could easily change these parameters if they were meaningful in their scenario. We also constrain the other parameters in our simulator so that the simulation is stable when the user is given control over these values. For example, it might be logical for the user to select between a weak, medium or strong promoter. In this scenario, the user should not be required to know the exact numerical values of their choice. Although more advanced users may wish to be able to input their own values. For this reason, our simulation uses sane defaults for the transcription rate, , which is analogous to promoter strength.
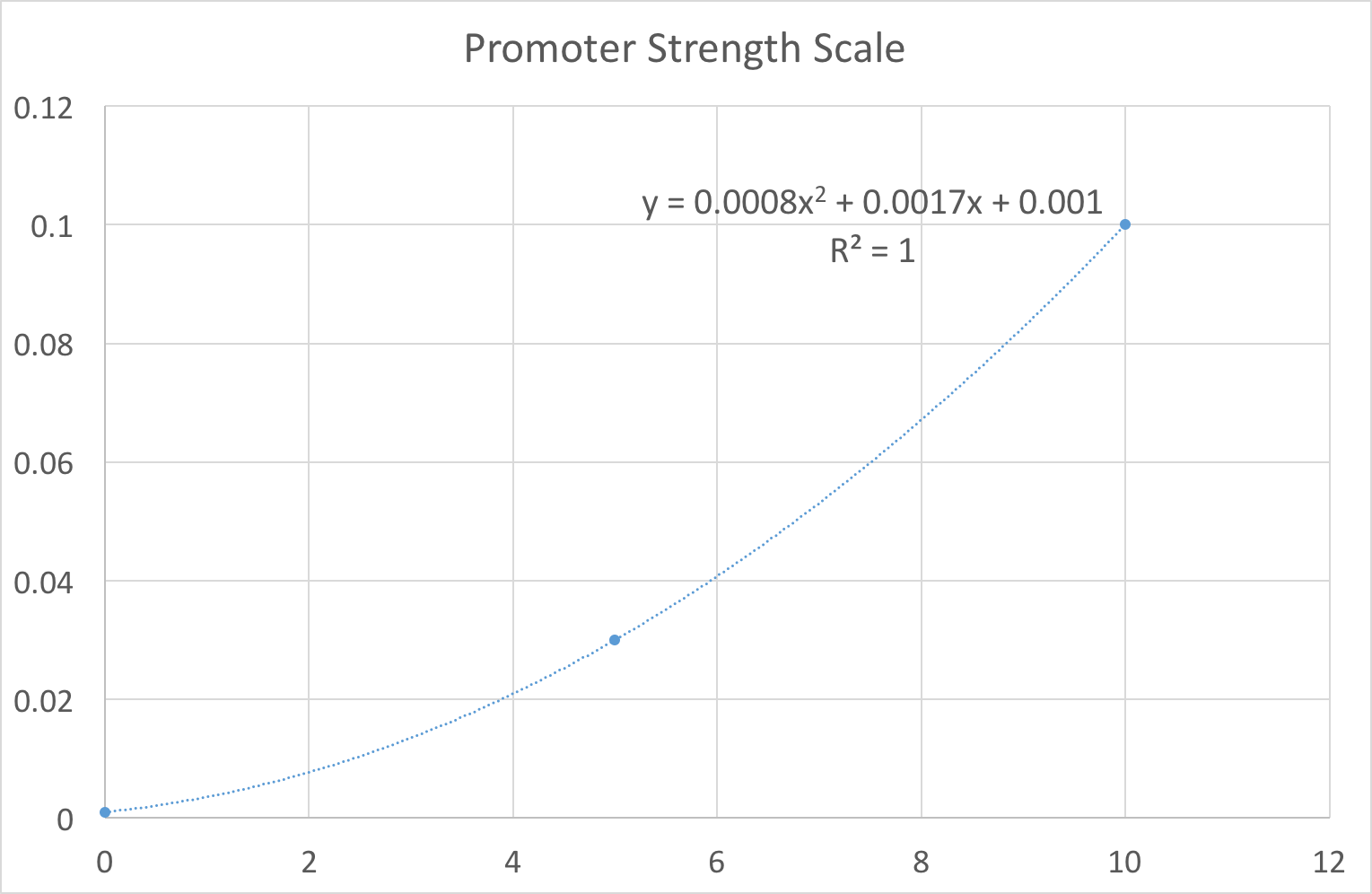
In our design work, users thought being constrained to these three options limited their ability to fully explore the biological system. For example, it would not allow the user to see the variation in gene product caused by slight noise in the promoter strength value, e.g. switching between 0.08 (supposed to be 0.03??) and 0.1 quickly. As our system is primarily a way of exploring the constructs we have built we wanted to limit the end user as much as possible. Therefore we set out plans to replace the notion of strong/medium/weak promoters with a more fine-grained scale. One that the user might control with a simple slider for example. To achieve this feat we fit our existing values using regression to a 10 point scale. This gave us an equation that we can use to map a user’s idea of ‘promoter strength’ to a transcription rate. As shown in Figure 1, the resulting equation was . Note that we deliberately ensured that to reflect the fact that there is always some background level transcription. In layman's terms, no promoter can be completely ‘switched off’.
The remainder of our simulator is built around these simple equations. The rest of this page will discuss how the other components were simulated using this technique and then integrated into the simulator. Our constructs were found to be easily adaptable to this form of model. However, it is not applicable in all situations. Our equations capture a simple ‘PRET’ (Promoter-Ribosome-Expression-Terminator) construct. As such it can be generalized to any number of protein expressions by having a tuple for each gene product in the model and separate counts of mRNA and protein.
Justification
We chose to build our simulator around a set of ODEs rather than using a more complex system that produced models in a format like CellML or SBML because it met our design goals. Primarily, this was ease of development and integration into the iGEM wiki system. Our main aim was to allow the simulator to be embedded in the wiki. In turn we felt that it was necessary to support the aims of the iGEM wiki system in preventing ‘bit-rot’, that is not having some aspects of our project hosted somewhere else where their uptime could not be guaranteed. This meant we needed a purely client side implementation and couldn’t ‘call out’ to other computers to perform the computation. When researching how we would construct our system we determined that the most straightforward of modelling our system in JavaScript without using any libraries or complex dependencies was too hard-code the ODEs.
Another aspect that makes the ODEs approach useful for us is that the level of abstraction they capture i.e., transcription and translation, corresponds directly with the level at which we design our BioBrick constructs. This makes it easy to translate between formats and allows for us to rapidly add new knowledge to the simulator as we develop our constructs. And vice versa, the simulations at this level translate directly to components in the system which we can control in our constructs. For example, we can directly influence the number of proteins in the cell by increasing the number of coding sequences on a construct. Whereas we can’t influence variables like the amount of free mRNA through our constructs alone.
There are some trade-offs to this approach. For instance, we lose access to tools that perform local and global sensitivity analysis, parameter estimation, etc., which limits the ‘depth’ of our models. We also lose out on interoperability with future software through interchange standards like SMBL. However, we believe that this doesn’t subtract from the quality of our models because their intended purpose is to encourage the user to think about the implications of interfacing bacteria with electronics rather than providing detailed models. In other words our present systems are not complex enough to warrant modelling at a lower level of abstraction in the simulator.
That is not to say that we didn’t also perform lower level modelling. We also used rule-based modelling in the BioNetGen language using the RuleBender graphical tool. This allowed us to capture our systems in more detail and to ‘sanity check’ the simplified models we use here in our simulator.