Difference between revisions of "Team:UBonn HBRS/Description/B Subtilis"
(Protein Purification 2/2) |
m |
||
| Line 150: | Line 150: | ||
eluting them. | eluting them. | ||
| − | === Protocol === | + | === Protocol === |
An overnight culture of 10 mL of medium with antibiotic | An overnight culture of 10 mL of medium with antibiotic | ||
(1mM, for T7-RNA-Polymerase, Amp) is grown at 37°C | (1mM, for T7-RNA-Polymerase, Amp) is grown at 37°C | ||
and 180 rpm. Inoculate Bacteria in 100ml LB and | and 180 rpm. Inoculate Bacteria in 100ml LB and | ||
| − | antibiotic (1mMol). The OD600 | + | antibiotic (1mMol). The OD600 is measured every 20 min until an OD600 of 0.5 is reached. Protein expression is |
| − | + | ||
| − | until an OD600 | + | |
| − | + | ||
then induced through the addition of IPTG (1mMol) and | then induced through the addition of IPTG (1mMol) and | ||
the samples are incubated for another 3-4 h. To control | the samples are incubated for another 3-4 h. To control | ||
Revision as of 22:55, 19 October 2016
![]()
B. Subtilis
General
Bacillus subtilis is a gram-positive bacterium which can be found almost everywhere, especially in soil. It can form a protective endospore to resist extreme environmental conditions.
We decided to work with this organism, because Bacillus subtilis is a bacteria, which naturally secretes proteins
and enzymes into its surrounding medium.
The goal was to utilise this attribute for enzyme production and create a “cell factory”. Instead of
lysating bacteria, it was desired to have a continuous production of enzymes in a liquid culture that
we can extract from the supernatant.
Repeated inoculations of new bacteria cultures are not necessary, because they will stay in the
stationary phase. Only the media has to be replaced.
Bacillus subtilis is classified as a GRAS-organism (general regarded as safe) by the U. S. Food &
Drug administration and widely used in S1 laboratories as a model organism. It is also present in
economy as probiotics in pharmacies or fungicide in agriculture.
Furthermore biotechnological applications already use Bacillus subtilis to synthesize enzymes.
Usage
For using our product we currently have two different approaches.
Either just using the supernatant to deink our paper fibers or make the time intensive and expensive
step of purifying our enzymes and then proceed with deinking the paper. For purification we
attached a His-tag to our enzymes to separate them from other components of the supernatant
with nickel columns.
In order to reach the project goals plasmids containing the needed genes had to be transformed into
the bacteria.
We worked with two different Bacillus subtilis strains which were donated by the department of
Pharmaceutical Biotechnology of the Greifswald University, Germany.
We got the wild type strain ATCC 6051 as well as the promising LS8P-D strain. The LS8P-D stain
is a derivate from the protease-deficient WB800 strain, where two additionally genes were deleted.
The lack of proteases is essential to secrete our enzymes without damaging them.
After trying out multiple protocols, we decided to accomplish transformation by electroporation.
The principle of electroporation relies on creating small holes in the cell wall of bacteria, by
sending a current through them. The plasmids enter the bacteria through the holes before the
organism repairs them.
The iGEM Team Freiburg and the iGEM Team Bonn collaborated in order to obtain working transformation protocols for Bacillus subtilis. The resulting manual “How to Bacillus subtilis” can be found on our site:
Protocol
To acquire electrocompetent Bacillus subtilis, the bacteria had to be cultivated overnight in LB medium. 500 μl of the preculture were added to each of the 3 flasks containing 20ml of competency medium (LB medium containing 0.5M Sorbitol) and the cultures were incubated at 37°C 250 rpm shaking till a OD600nm between 0.5 and 0.7/ml was reached. Glycine was added as a weakening agent, replacing the alanine in the cell wall leading to loosen it, for total concentrations of 1%, 1,25% and 1,5% in the different flasks. This was followed by a 60min incubation. The cells were cooled on ice for 15min and were pelleted by centrifuging at 8500 rpm for 10min at 4°C. The supernatant was removed and the cell pellet was washed repeatedly through a circle of resuspending, pelleting and discarding of supernatant. The washing steps were performed with washing buffer (0.5M Sorbitol, 0.5M Mannitol and 10% Glycerol). Finally all pellets were combined and resuspended in washing buffer. Aliquots were made and were frozen in liquid nitrogen before being stored at -80°C.
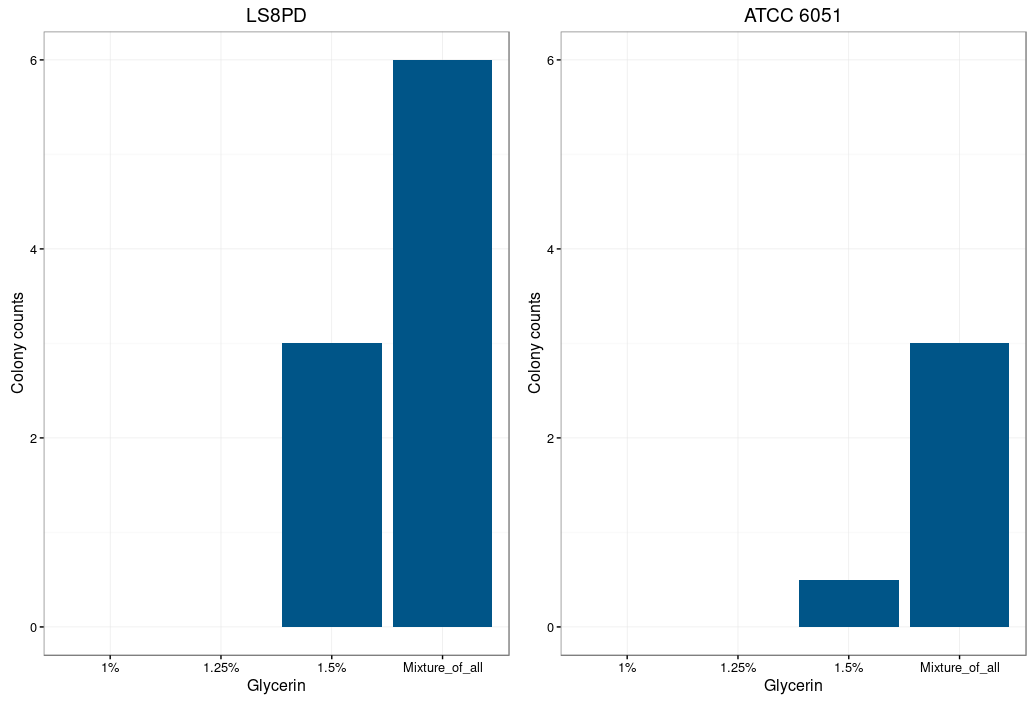
Glycine is supposed to act as a weakening agent, by replacing the alanine in the cell wall and thereby loosening it 2.
Electroporation worked best with a higher glycine concentration. However a too high concentration prevents bacterial growth and could also be toxic. The highest colony number was measured by using a mixture of all three glycine concentrations (1%,1.25% and 1.5%) for the preparation of electrocompetent cells.
In order to transform the microorganisms, an electroporator was used with the fitting cuvettes.
For the electroporation a mix out of plasmid DNA and competent bacteria was prepared with a volume of
60 μl containing 10ng of DNA per μl. The mixture was cooled on ice for 10 min along with the
electroporation cuvette and was electroporated at 2,1 kV. 1 ml of competency medium was
used to flush the cells out of the cuvette and the culture was incubated at 37°C 250 rpm shacking for
about 3h before it was plated on LB agar containing kanamycin.
| DNA-concentration (ng/μl) | Number of colonies |
|---|---|
| 10 | 11 |
| 5 | 3 |
| 1 | 1 |
| 0 | 0 |
The table shows the number of formed Bacillus subtilis colonies after transformation using electroporation with varying DNA-concentrations. It shows that a higher amount of DNA (10 ng/μl) leads to a higher number of colonies
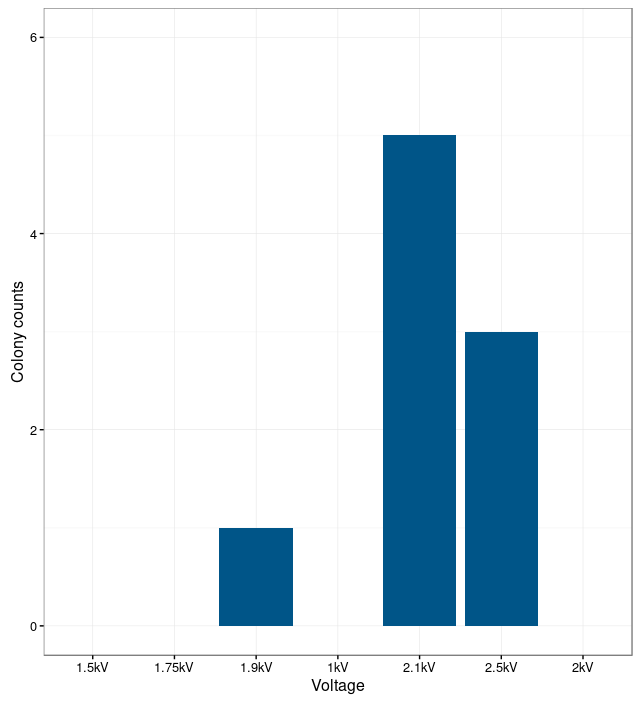
In order to find the optimal electroporation current, electroporation was performed with different voltages. The electroporation was unsuccessful at low currents. The bacteria were not able to take up the plasmids. With too high currents, the bacteria were lysed. The results show that the optimal current for electroporation is at 2.1 kV. At this current the number of successfully transformed bacteria was the highest.
Protein Purification
The aim of this project part is to express and secrete our proteins in Bacillus subtilis and purify them. The enzymes used in this project are equipped with a C- terminal polyhistidine-tag (his-tag). A his-tag is a sequence of at least 6 histidine residues, giving the protein of interest an affinity for nickel. Through the complexed nickel ions in Ni-NTA (nitrilotriacetic acid) columns, the proteins containing his-tags can be isolated from other proteins. Specific detection methods via small molecules or antibodies for his-tags also exist, allowing for the simple validation of enzyme expression.
The first step in the protein purification procedure, is to cultivate the bacteria containing the plasmid with the protein to be expressed. Protein expression is then induced through the addition of IPTG (Isopropyl-β-D- thiogalactopyranosid). After further growth for 3-4 hours, the cells are harvested and lysed. The lysed cells are then transferred to Ni-NTA columns, where the his- tagged proteins bind to nickel bearing agarose beads, separating them from the rest of the cellular material. Next the columns are washed to remove all irrelevant material. Lastly the his-tagged proteins are eluted from the beads through the addition of Imidazole (C3N2H4)o. Imidazole, a ring structure also found in the histidine structure, competes with this molecule for binding to the nickel beads thus displacing the his-tagged proteins and eluting them.
Protocol
An overnight culture of 10 mL of medium with antibiotic (1mM, for T7-RNA-Polymerase, Amp) is grown at 37°C and 180 rpm. Inoculate Bacteria in 100ml LB and antibiotic (1mMol). The OD600 is measured every 20 min until an OD600 of 0.5 is reached. Protein expression is then induced through the addition of IPTG (1mMol) and the samples are incubated for another 3-4 h. To control your results it is wise to collect a sample before and after IPTG induction to compare the content of your protein of interest. The bacteria are pelleted at 3000g at 4°C for 30 min. The pellets can either be stored at -20°C or you can immediately proceed with lysing. The bacteria pellets are lysed through the resuspension in 25 mL of lysis buffer prepared according to table 1 and followed by sonification with the settings listed in table 3.
| 50 mM Tris |
| 300 mM NaCL |
| 10% Glycerol |
| 20 mM Imidiazole |
| Ph7.8 |
| 1 min 40%/cont |
| 1 min 70%/cycle 30 |
| 5 min incubation on ice |
| 1 min 70%/ cycle 30 |
The samples are then centrifuged at 3000g at 4°C for
one hour. In this time the agarose beads are prepared.
For each sample you need 1.2 mL Ni-NTA agarose
beads. They are centrifuged for 2 min at 1000rpm and
the supernatant is discarded. The beads are washed
twice with 1.5 mL lysis buffer. After each washing step
the beads are centrifuged again for 2 min at 1000 rpm
and the supernatant is discarded. After washing the
beads are resuspended in 1 mL lysis buffer and stored
on ice. The cleared lysates are distributed into 15 mL
falcons and the Ni-NTA beads are added. The samples
are then incubated at 4°C for 30min to an hour using an
overhead tumbler. The samples are then applied to
large Biorad columns and the flowthrough is
collected. The beads are washed 4x with 5 mL of lysis
buffer each. The wash fractions are collected for later
analysis. Finally, the proteins are eluted through the
addition of 2 x 1.5 mL of elution buffer prepared as
described in table 4. The eluate is collected and kept on
ice. To further purify the protein, the eluate is transferred
to dialysis tubing and dialyzed overnight at 4°C in
dialysis buffer (refer to table 5).
| 50 mM Tris |
| 300 mM NaCl |
| 10% Glycerol |
| 250 mM Imidazole |
| pH 7.8 |
| 500 mM Tris |
| 100 mM NaCl |
| 0.5mM PMSF |
| Ph 8.0 |
All fractions of protein purification, i.e. flowthrough, wash
fraction and eluate are analysed via an SDS page gel.
Visualization
After purification, a SDS page gel is used to analyze the amount of his-tagged proteins in each fraction of the purification procedure, as well as to confirm the expression of the protein. First the bands of the gel are analyzed on a coomassie gel, to confirm the presence of proteins (compare Fig. 3). The his-tagged protein is then specifically visualized via InVisionTM His-Tag In- gel staining. Invision staining results in fluorescent bands that can be detected using UV light (compare Fig. 4). We performed this protocol according to manufacturer's instructions (refer to InVisionTM His-tag In-gel Stain manual, Macherey and Nagel).
Due to the weight variation (22,6 kDA – 107 kDA) of our Enzymes, we used a 12% SDS Page-Gel. The gels were prepared according to table 6. The gels were run at 150V 500mA 30W for 1h.
| Seperating gel 8ml : |
| Aqua dest (ml) 2,64 |
| 30% Acrylamide (ml) 3,2 |
| Tris 1,5 M, pH8,8 (ml) 2 |
| SDS 10% (ul) 80 |
| Aps 10%(ul) 80 |
| TEMED (ul) 8 |
| Stacking gel (5%) 4ml : |
| Aqua dest (ml) 1,36 |
| 30% Acrylamide (ml) 0,34 |
| Tris 1,5 M, pH8,8 (ml) 0,26 |
| SDS 10% (ul) 20 |
| Aps 10%(ul) 20 |
| TEMED (ul) 2 |
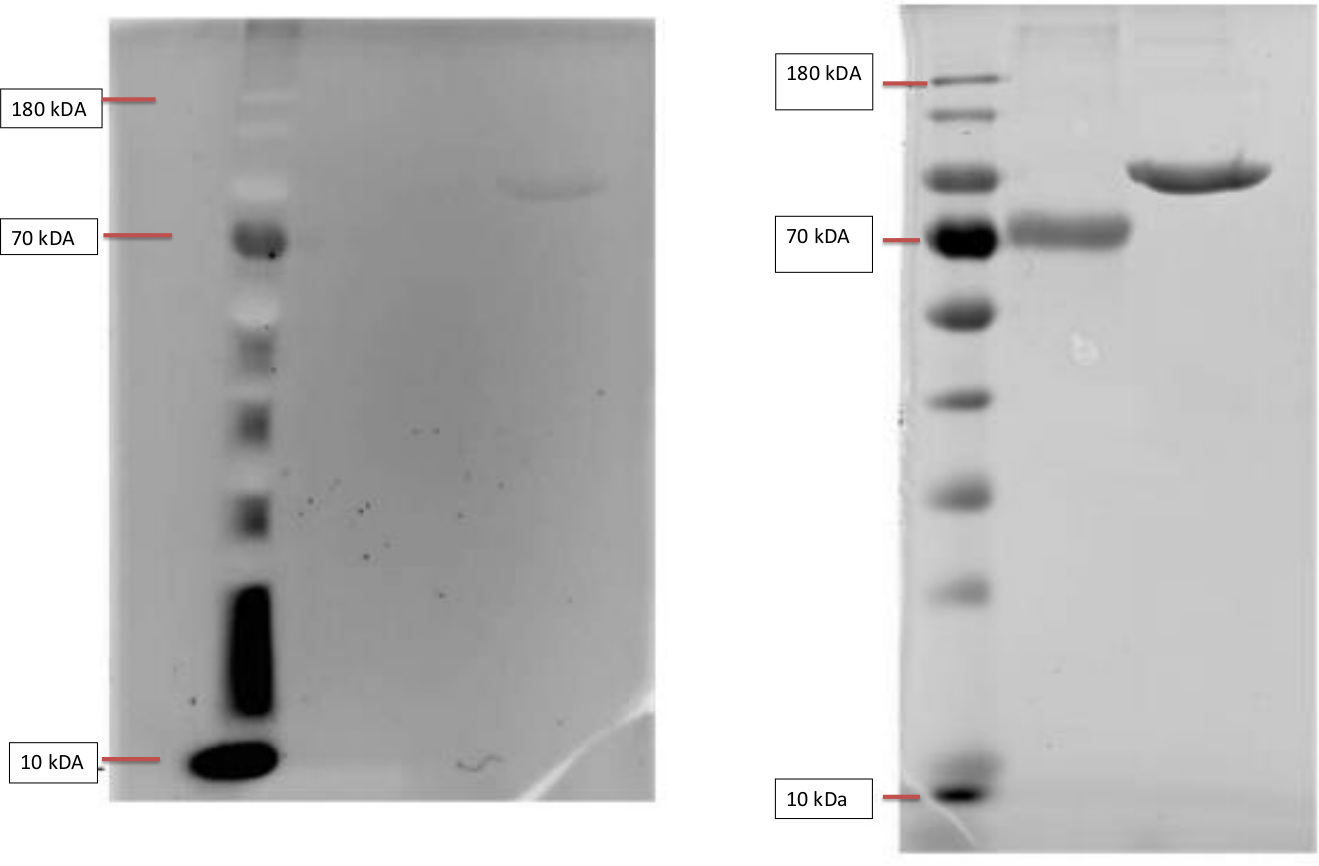
Left: InVisionTM His-tag In-gel Staining of Ladder, BSA and T7-RNA-polymerase.
The Invision Stain was imaged with a Gel doc. The samples were loaded onto the gel in following order, BSA, BSA and T7. The BSA serves as a negative control, as it lacks a his- tag and should not show flourescence. The T7-RNA-Polymerase however contains a his-tag and is thusly visible as a band in the third column.
Right: Coomassiestain of ladder, BSA and T7-RNA-polymerase.
The figure shows a test of our SDS-Protocol and Coomassie staining. The gel shows the bands of our negative control non his-tag protein BSA (Bovine serum albumin) with a weight of 66.5 k in comparison to our positive his-tag protein T7-RNA-Polymerase with a weight of 99kDA.
Due to the lack of time, it was not possible to analyze the
expression of our proteins.
1: http://www.fda.gov/Food/IngredientsPackagingLabeling/GRAS/
2: Zhi Zhang et al.: Development of an Efficient Electroporation Method for Iturin A-Producing Bacillus subtilis ZK