Team:XMU-China/Proof
Proof Of Concept
Verities never lie.
Experiment
Entire circuits test
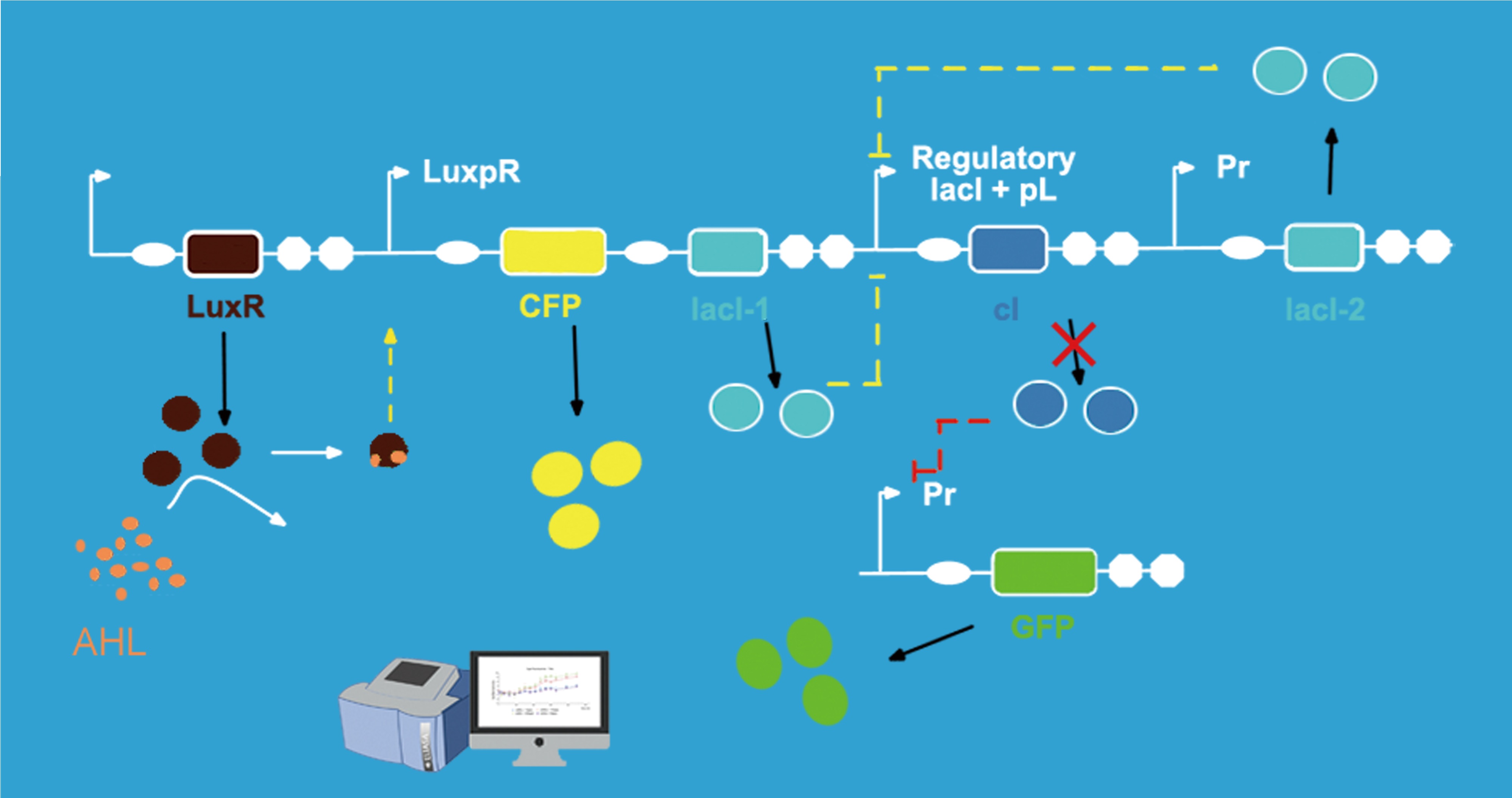
For the fluorescence is easier to measure, in our experiment, we used the cfp gene and gfp gene to replace the responder and lysis respectively when constructing the gene circuit in order to test the circuit more conveniently. Also, the strength of the fluorescence can stand for the strength of the corresponding genes' expression.
When AHL around the engineered bacteria is at low concentration, AHL can't be sensed by the Lux sensor part, and the cI gene is expressed at the wild-type levels. While lacI-2 and GFP are repressed since the CI protein is at high concentration. When AHL around the engineered bacteria reach a certain concentration, AHL can be detected by the sensor part. So the promoter LuxpR is activated, CFP and lacI-1 express. So the CFP protein can be tested by fluorescence measurement. And the lacI-1' product-LacR protein can inhibit the expression of cI gene. As a result, lacI-2 and GFP can express. The LacI-2' product can further repress cI gene's expression, and GFP protein will be measured by fluorescence detection to represent the level of lysis gene expression. The time interval of firstly detecting CFP' fluorescence and firstly detecting GFP' fluorescence can represent the delay time which the switch give for responder expression.
Result
1.AHL Gradient Induction
The expression of our circuits could be induced by AHL. When AHL is sensed by the sensor part, the promoter LuxPR is activated, CFP and GFP are expressed.
We found that the concentration of AHL could have an effect on the expression of our circuits, in order to find out the relation, we decided to make an AHL gradient induction for our gene circuits.
The E. coli containing our circuits were divided into two groups: experimental group with different concentrations of AHL and blank control group without AHL. Then we measured OD600 of the E. coli and the fluorescence intensity of CFP and GFP for 900 minutes. The OD600 showed the growth trend of the E. coli. The fluorescence intensity data and time on the AHL of different concentrations show the relationship of AHL to the expression of our circuits.
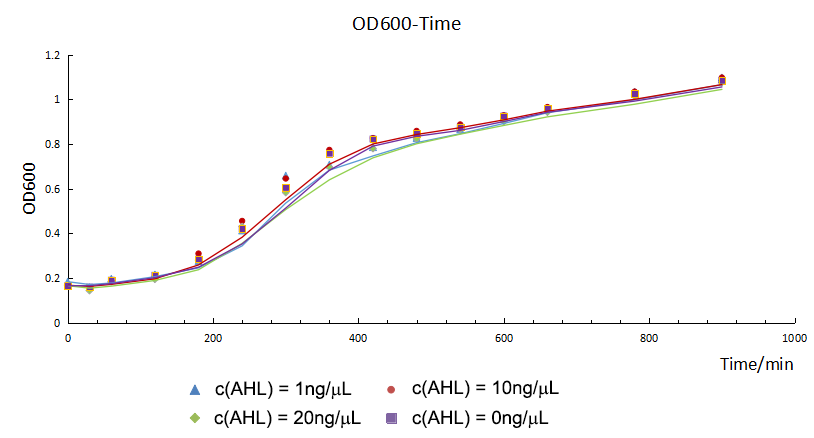
The growth curves were similar. It was suggested that they had the same growth trend and the difference of fluorescence intensity was mainly from the expression of circuits.
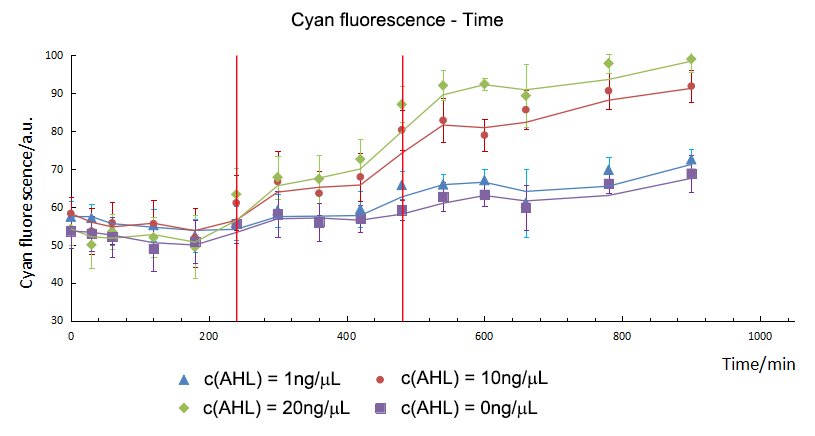
AHL would influenced the expression of CFP directly. The higher the concentration of AHL was, the more CFP expressed in a range from 1ng/ μL to 20ng/μL. In addition, the expression time of CFP was different. The fluorescence intensity between the experimental groups and control group were compared with independent t-test. It showed that CFP in 10ng/ μL and 20ng/μL group started to express in the 240th minute but it started to express in the 480th minute in 1ng/μL group.
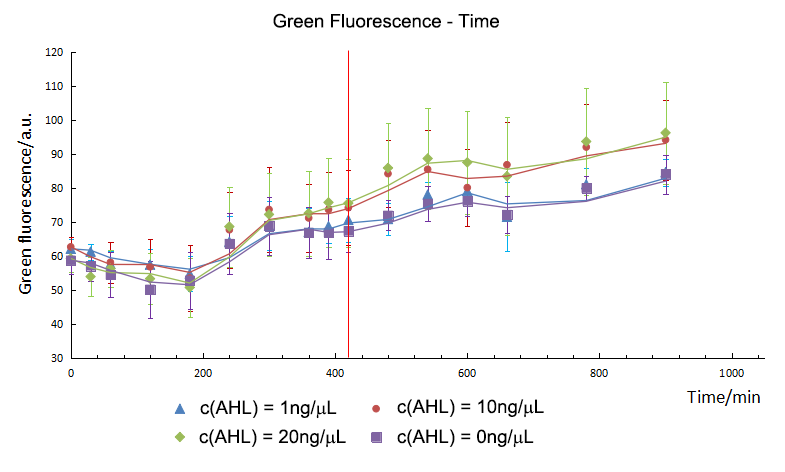
The concentration of AHL didn’t influence the expression of GFP directly. The GFP in 10ng/μL and 20ng/μL group started to express in the 420th minute and it had no significant difference in expression level. The GFP didn’t express in 1ng/μL group.
2. 2.The Delayed Effect
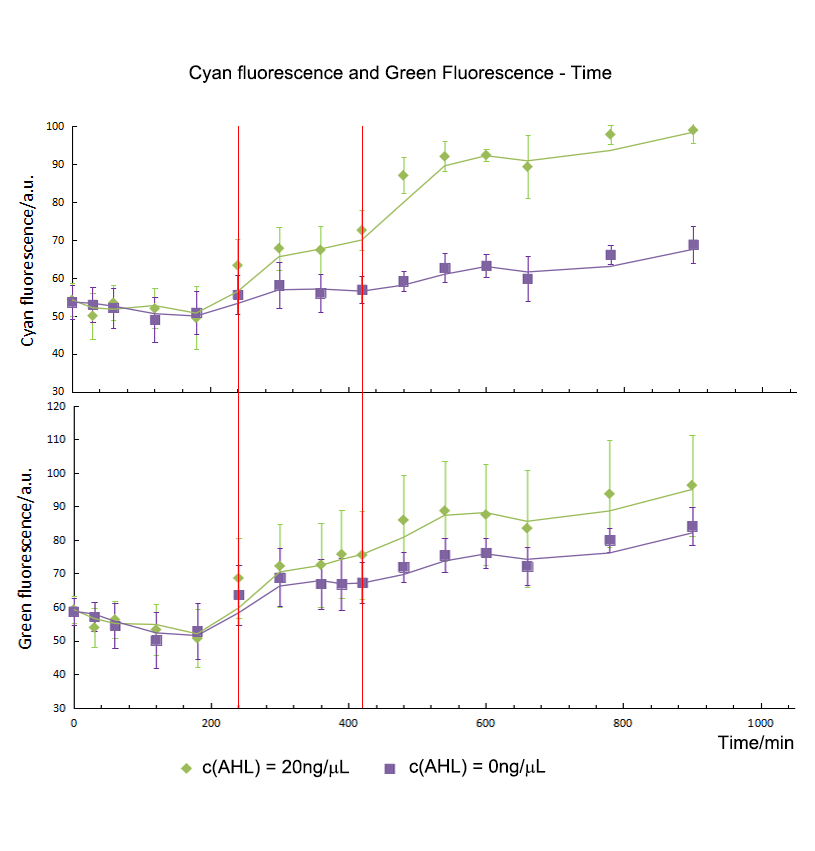
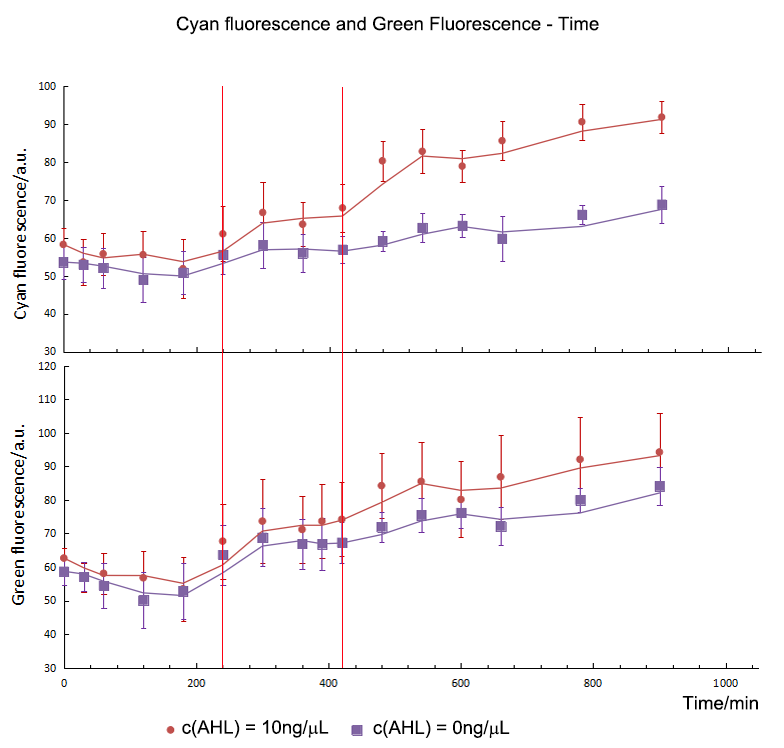
T-test showed that the CFP and GFP in experimental group started to express in the 240th minute and 420th minute separately on the AHL of 20ng/μL. The expression time of GFP was delayed to 180mins post CFP. The delay time is the same on the AHL of 10ng/μL.
Responder test
Against the backdrop of today’s widely spread of drug-resistant bacteria, small regulatory RNA (sRNA) offers a new clinical therapy approach to repress the target drug-resistant genes and then it can enhance the effect of antibiotic in the clinical. Here we designed synthetic sRNA to target the antibiotic resistance gene chloramphenicol acetyltransferase (CAT) to generate representative data and to quantify knockdown effectiveness.
We designed and synthetized RNA silencing cassettes (Table 1) and constructed it into pSB1AC3, an official plasmid which contained ampicillin resistance gene and chloramphenicol resistance gene. Also, we used pSB1AC3+mRFP as control.
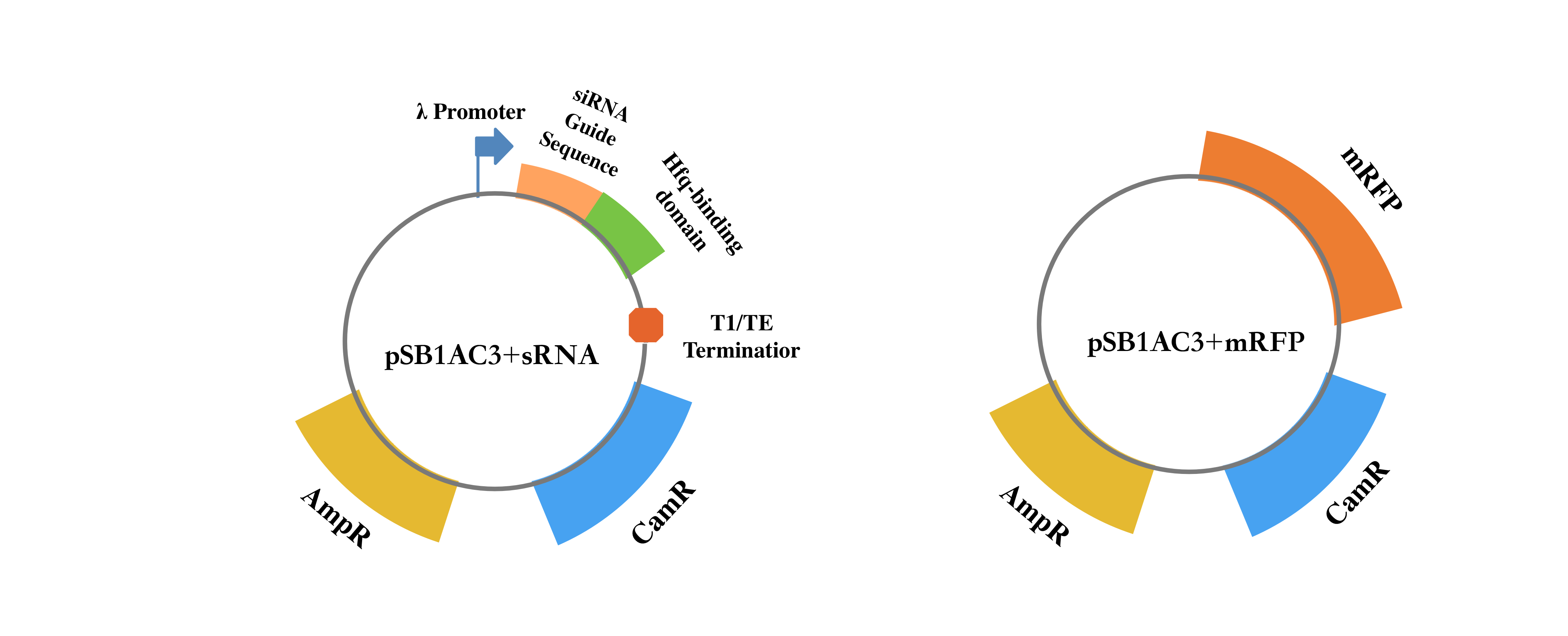
| Part | Sequence |
|---|---|
| pR promoter | TAACACCGTGCGTGTTGACTATTTTACCTCTGGCGGTGATAATGGTTGC |
| GUIDE sequence | ATATCCAGTGATTTTTTTCTCCAT |
| Hfq binding domain | TTTCTGTTGGGCCATTGCATTGCCACTGATTTTCCAACATATAAAAAGA CAAGCCCGAACAGTCGTCCGGGCTTTTTTTCTCGAG |
| T1/TE terminator | TTTCGTTTTATCTGTTTTTGTCGGTGAACGCTCTCTACTAGAGTCACACT GGCTCACCTTCGGGTGGGCCTTTCTGCGTTTATA |
We transferred the plasmids into chemical competent cell (DH5α) to detect the knockdown effectiveness through the phenotype of survival index. We continuously measured the OD600 for 9 hours, it showed the growth trend of the bacteria with the sRNA cassettes and without.
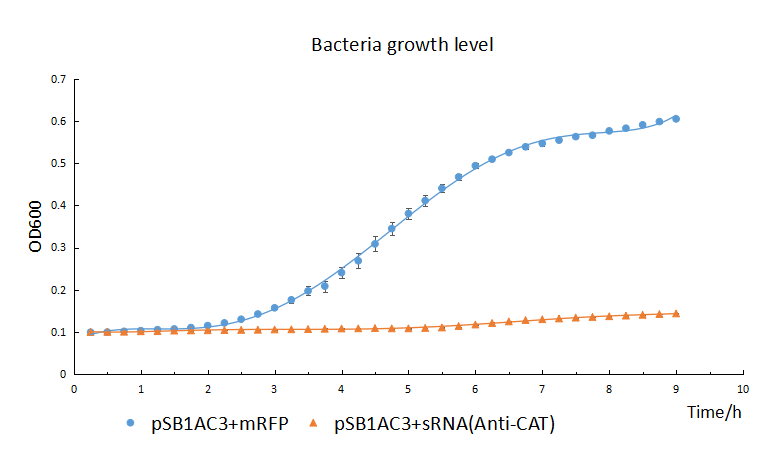
The growth of chemical competent cells(DH5α) with pSB1AC3+sRNA is significant repressed by the chloramphenicol (50μg/mL) compared with the control with pSB1AC3+mRFP (The blue line). It indicated the sRNA’s highly knockdown effectiveness.
The result suggested the growth of the bacteria which contained the sRNA cassettes significantly were repressed by the chloramphenicol (50μg/mL).
Model
Abstract: E. coli has been modified in order to produce small interfering RNA molecules (siRNA) to attack the resistance genes that become available for nearby bacteria, when a resistant strain goes through lysis. However, a regulation system should be applied so the productive cells do not become a problem. A circuit has been implemented, this circuit regulates the transcription of said siRNAs among the production of toxins that could lead to death of any nearby cell, including the siRNA producers themselves. Taking in consideration all these elements a mathematical model using differential equations has been designed to approach the number of viable cells within the toxic environment once the circuit has been activated.
Keywords: siRNA, toxin, mRNA, transcription, translation, exponential
Rate of production of toxin mRNA and toxin molecules
The toxins are produced through a gene in the circuit, so the production of toxin depending on the translation of the mRNA produces, which also depends on transcription variables. A differential equation was taken in consideration in order to measure the mRNA rate production.
\(K\) represents the transcription rate, which is a constant parameter, in molecules of RNA per second, \(D_{r}\) is the degradation rate expressed in units per seconds as it depends on how much mRNA is actually present on the cell, expressed by the variable “\(r\)” in units of molecules of mRNA produced.
Afterwards, the original mRNA production equation was obtained through the integration of the differential equation by linear integration.
Establishing a new condition \(r(0)=R_{0}\) then we can complete the system. Substituting we obtain:
We finally obtain the equation for mRNA synthesis:
To simplify the model we assume that each mRNA molecule will be transcribed in order to produce one molecule of protein. This means that each molecule of mRNA produced is equivalent to one molecule of active toxin that in certain concentration can kill bacteria. We assume a fast degradation of mRNA inside the system and almost no significant dispersion of the molecules, so this ratio can be respected.
Productive cells inside the system
The number of E. coli cells within the system can not be considered as a constant, neither as a linear function due to the exponential nature of cellular division. Given normal growth of the cell culture, it is possible to describe the number of E. coli cells as a function of time. Nevertheless, the E. coli concerning the model produces toxins that alter the number of E. coli cells itself, also there is a natural decrease in the number of bacteria caused by cellular death, resulting in the addition of new terms to the traditional equation. An equation considering all the previous factors is shown below.
Where:
\(C_{0}\): Initial population [=] cells
\(C\): Total productive cells [=] cells
\(D\): Cell fraction ( \(C_{0}e^{Gt}(1-D)\) ) that dies naturally [=] adimensional < 1
\(G\): Rate of cells that are born [=] 1/second
\(M\): Fraction of viable cells ( \(C_{0}e^{Gt}(1-D)\) ) that lyse due to toxins [=] adimensional < 1
\(D\) and \(M\) are not related, therefore they are independent. It is also important to notice that we assumed that the number of toxins equals the number of mRNAs transcribed, so our previous defined function \(r(t)\) can help us to determine the constant \(M\).
where \(X\) is the critical toxin production rate. Also in order to determine the \(M\) term we need other variables.
\(M=\frac{T r(t)}{(1-D)C_{0}e^{Gt}}\), where:
\(r(t)\) = toxins produced in time \(t\) [=] (molecule of toxin)
\(T\) = cells that die by unit of toxin [=] (cells / molecule of toxin)
\(M\) = dead cells / total viable cells [=] adimensional
The coupled equations yield the following final modelling:
Discussion
When doing our experiment, we didn’t test M13 gene and T4 endolysin gene directly. The reporter gene cfp and gfp were chosed as the alternate to construct our whole circuit. It’s advisable to use the fluorescent protein gene, for fluorescence is easy to measure. After we add AHL into the culture medium, it is convenient to determine when the responder expresses, for that we only need to take out a little bacteria liquid and test its fluorescence. Also, the delay time of the toggle switch becomes simple to measure, because we just determine the time interval of firstly detecting CFP’ fluorescence and firstly detecting GFP’ fluorescence.
As for the test of the sRNA silencing cassettes, we put it into the backbone pSB1AC3 which contained ampicillin resistance gene and chloramphenicol resistance gene and transferred the plasmids into chemical competent cell (DH5&alpha). If the sRNA silencing cassettes works, the cell can’t resist chloramphenicol. To see the effect of sRNA silencing cassettes, all we need is just to measure the OD600, and know the growth trend of the bacteria after we put chloramphenicol into the bacteria culture medium.
Reference
[1] Bernheim, A. G., Libis, V. K., Lindner, A. B. & Wintermute, E. H. Phage-mediated Delivery of Targeted sRNA Constructs to Knock Down Gene Expression in E. coli. J. Vis. Exp. 109, 1–10(2016).
[2] Kobayashi, H., Kærn, M. & Araki, M. et al. Programmable cells: Interfacing natural and engineered gene networks. PNAS 101, 8414-8419(2004).


CONTACT US
 https://www.facebook.com/IGEM-XMU-China-1067638406629389
https://www.facebook.com/IGEM-XMU-China-1067638406629389 https://www.twitter.com/Amoy_igem
https://www.twitter.com/Amoy_igem igemxmu@gmail.com
igemxmu@gmail.com
Name: XMU-China School: Xiamen University
Address: Xiamen University, No. 422, Siming South Road, Xiamen, Fujian, P. R. China 361005
CONTACT US
https://www.facebook.com/IGEM-XMU-China-1067638406629389
https://www.twitter.com/Amoy_igem
igemxmu@gmail.com
Name: XMU-China School: Xiamen University
Address: Xiamen University, No. 422, Siming South Road, Xiamen, Fujian, P. R. China 361005