Team:Cambridge-JIC/Notebook

NOTEBOOK
27th June
Entire team assembled for the first time in the Department of Plant Sciences. Had some fun in building sticky note tower (the tallest one was 2.5 meters, built in one hour without previous preparation!) Various coffees and teas are available in the department, very planty.
In the first week we had lectures and discussion on synthetic biology, photosynthesis, DNA assembly and genetic engineering on plants. We also practiced using Geneious software and visited the botanic garden. Lots of brainstorming creates tonnes of ideas about ways to save the world using plants in 10 weeks.
1st July
By the end of the week we have a long list of ideas but nothing too concrete so that we could assess feasibility. We have made a big progress in getting the team together, communicating biology to engineers, finding the common language. By monday we have a challenge - to everybody come up with a well elaborated idea about what to do.
4th July
Bio-plasters
We got all really excited for bioplasters. Lucie has found two cool articles where they have introduced Chlamydomonas into biomaterials to produce oxygen. It worked nicely and caused no immune reaction but they haven't modifies Chlamy to do something cool. We could do that! The group has elaborated on it further but then we realised it would be hard to find supervisors for that and there might be issues with working with mammalian material. We had a presentation from everybody about our ideas - Geoff about light-producing algae, Mie about environment cleaning and biohouses, Jake about growth facility, etc.
5th July
Finding our biological direction: Homoplasmic transformation
After a meeting with advisors we came to reconsider our idea of establishing a plant toolbox opening the Plant Track and especially chloroplasts to iGEM and science. And we found a very exciting goal - to make a tool which would propagate our constructs into all the plastid DNA copies quickly and efficiently using CRISPR/Cas9. No more tedious selection rounds for 2-3 months!
In the following days we discussed with the experts in the Plant Sciences Department and dived into the literature on chloroplast transformation.
6th July
Promotion and Marketing
8th July
Visiting Cambridge Consultants
All members were invited to Cambridge Consultants. We discussed about the project, visited their laboratory and had a delightful lunch in the cafe. They are really keen to give us advice! In the afternoon we have established some strategic roles - who will manage the team, work on graphical design, wiki programming, content, etc.
10th July
Scientific 'pool'
Still swimming in the scientific literature and trying to get as much help from our busy advisors as possible. Last week we aimed for sending all the sequences for synthesis today. And do everything at once. But over the time we realise that eating an elephant requires cutting it into smaller pieces. We need to test everything to know what went wrong, we plan controls for everything and design experiments which will teach us even if the result is negative.
But we are still thinking far ahead and considering actually introducing some gene of interest by our finished system, if it works. We spend a lot of time researching into edible vaccines and virus-like particles which could have an impact in the field and it's really exciting!
11th July - 13th July
Co-lab workshop
Lovely French students were were coming down to Cambridge to give us a workshop about connecting synthetic biology with design. An opportunity not to be missed! We joined some keen designers as well as people with other backgrounds and together we were working in the lab, had an etnographic workshop, had interesting lectures and were looking for a common language. We got a lot of interesting inputs for our human practices and got to design a few human practices projects happening later. For example we want to make discussion workshops in between students interested in synthetic biology, philosophy and journalism to bring them together to share thoughts. Hopefully we'll also go to schools to educate about synthetic biology.
15th July
Wet-lab setup
18th July
Protocols
We intensely worked on some easy protocols - tried our miniprep for amplifying our desired DNA in bacteria, culturing am bulking up our Chlamydomonas as well as preparing general and antibiotic plates for them. We have set up an experiment testing how well Chlamydomonas grows under different conditions - in the dark on TAP, in the light and in the light with CO2 concentration elevated to 5%. Thanks Dr Moritz Meyer for massive help!
19th July
PLASMID DESIGN UPDATE
Working further on our plasmids made of DNA parts. We have so far considered many different designs. Initially we planned to have two plasmids - one with the Cas9 regulated by a regulatable promoter which would be only transiently expressed. The second plasmid with the gRNA under one promoter and a separate one promoter for our markers and the gene of interest. We considered luciferase of fluorescent proteins as our markers. We were struggling to find a sharp promoter and terminator functional in chloroplasts so that our gRNA has the desired length and sequence in the end. We even considered ribozymes which are capable of self-splicing of the gRNA and used in mammals.


21st July
Parts for synthesis?
Today we thought our synthesis sequences were done, just needed final checks. Haha, had no idea how many major changes of the whole technique will come before we send them off. But with every change we learn more and more, nothing is a lost time!
We are also in constant contact with the lab of Prof Saul Purton at UCL who is giving us amazing advice from experience with working on this system and even some material his lab produced. He has been amazingly helpful especially when we need to decide on alternative parts - they know what usually works in Chlamydomonas chloroplasts!
22th -23th July
iGEM + Biodesign NightScience & iGAMER
During the Friday and Saturday we visited Paris for a cool conference about design of biological systems, DIY hardware, community labs and workshops, games teaching science, we have given a talk about our project. Were networking with many people from the whole Europe showing us also perspectives of designers. We made friends with Paris Bettencourt iGEM team and established a collaboration.
25th - 27th July
Open Plant Forum conference in Norwich
We went to Norwich to participate a very inspiring conference on synthetic biology in plants. The two days were packed with lectures and panel discussions from scientists from our Plants Department, Norwich (JIC and other institutes as well) and from some commercial companies. The Open Plant initiative aims to promote sharing of knowledge, working materials - physical as well as DNA sequences which would pave the way towards bigger inventions in science and industry much the same way standardised basic parts do in engineering.
During all the lunch and tea breaks as well as evenings we worked very hard on all our sequences, were chasing all the amazing scientists to discuss our project and get advice. It was amazingly productive and we thank a lot to all the people moving our project further, especially Dr Nicola Patron, Dr Tom Knight, Dr Steven Burgess and Ms Kher Xing Chan (Cindy)!
28th July
Preparing lawns of cells for Biolistics tomorrow. Collecting protocols.
We hoped to send out our sequences but there are more and more problems appearing with their nature (low GC content, length, etc.) so we keep working on them.
Got some results from our growth experiment. Our algae grow certainly the best in light and with elevated CO2 concentration in the air.
An article about us came out on Cambridge Consultants blog!
29th July
FINALLY SENDING OFF OUR SEQUENCES! (PLASMID DESIGN UPDATE VOL. 2)
Some had too high AT content and repeats to be synthesised (primers surprisingly :D ) but we will try to amplify them from the chloroplast genome. Our plasmid design had at least 10 major changes of direction and was evolving quicker than some of us were able to track! But planning is important, right?
Some of us have spent the afternoon learning biolistics - transforming our cells with fluorescent proteins
PLASMID DESIGN UPDATE VOL.2:
MARKERS
- First we have abandoned the idea of marking our genes with luciferase as that would not give us quick and easily detectable response. Fluorescent proteins should be more reliable although there are troubles with expressing them in Chlamydomonas chloroplasts - well, something for us to troubleshoot with trying our 18 possible FPs!
SAFETY - CAS9 REGULATION AND WAYS OF EXPRESSION
- We are very concerned with safety as our design may resemble a gene drive, we want to make sure we are definitely not making one. We were considering Lac regulatable promoters, light regulated ones and also Nac2. Nac2 is still a system we consider to try - we could switch off the expression of Cas9 when needed and regulate it in case it's toxic in higher amounts by concentration of thiamin. But it is actually in the UTR and regulated by a nuclear gene which regulates a photosynthetic gene. So we need to obtain a strain which has been designed to have these pathways independent, we'd need to optimise concentrations of thyamin and design more constructs.!
- We also considered another level of safety by introducing another gRNA which would make the Cas9 to cleave itself after homoplasmy has been achieved. However as NHEJ does not exist in chloroplasts, we would also need to supply the template. Still an option! But at the moment we plan to introduce Cas9 cassette either transiently or just into one copy of DNA, not homoplasmic (we plan to try both), let it drive the propagation of our genes of interest and then be lost when we remove the selection pressure of antibiotics. A semi-stable type of integration was decided as an alternative to transient because transient may not produce enough product and be list too quickly.
- We are also considering delivery of the preassembled complex of gRNA and Cas9 or its parts into chloroplast by biolistics, although it might be beyond our time-scope
- Because of safety our Cas9 and gRNA were initially separated either physically or one of them was outside the homology regions so that it won't be integrated. It was because we had only one insertion site and corresponding homologies. In the current design the Cas9 and gRNA are in one construct but gRNA is targeting another region on the chloroplast genome (so that it drives expression).
- Our polycistronic design of cassette might be simpler and produce shorter DNA fragments but maybe complicated for cell processing so we decided to put separate promoters for gRNA, antibiotic resistance and Cas9 fused with FP
ACTIVITY OF CAS9
- We had troubles with assessing that our Cas9 is active as one of the first steps. It would be very elegant to let it drive propagation of its own cassette with markers (or changing colours of algae already transformed with a fluorescent protein), making the strain less viable in field but the same in the lab (eg. by nutrient utilisation defficiency). The best solution would be restoring functionality of some mutant strain by the cassette. But we want to avoid a gene drive at all costs. We have also considered surveyor assay. So we are still looking for ideal solutions, probably by tracking Cas9 cuts by qPCR. (or do we have a solution?)
- Our Cas9 is now fused with the fluorescent protein. It means each molecule of the FP corresponds to a molecule of Cas9. And we can supply an interesting part into the registry
TYPES OF CAS9
- We were also considering different versions of Cas9 - from different bacteria, expressed from nucleus targeted to chloroplast (unluckily it is probably toxic in the cytoplasm), transiently or stably expressed, and for long we were considering using nickase (which makes just single-stranded breaks and so pair of gRNAs is used to reduce off target activity by around hundreds of folds). But then we learned the toxicity is more likely because of sucking RNAs rather than cutting off-target.
WORKING SYSTEM
- We considered prototyping experiments on isolated chloroplasts to speed them up but at the moment we plan to transform chloroplasts in the whole Chlamydomonas
INSERTION SITES
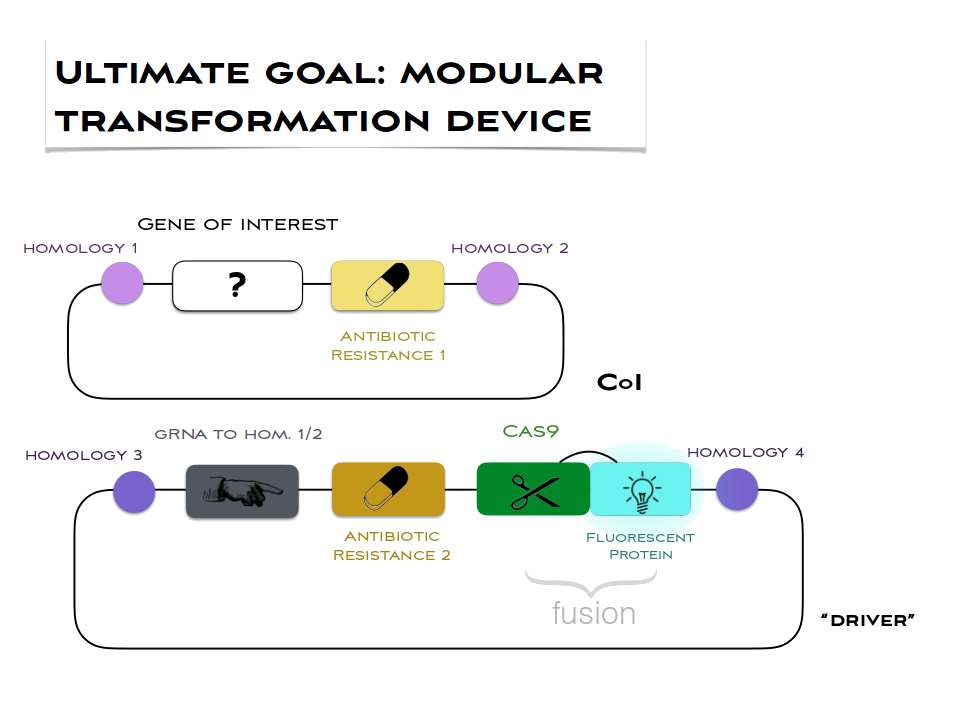
- This design also enables any researcher with a prepared cassette for insertion to work with our system just by changing the gRNA (20 nucleotides) to correspond to the desired site of insetrion. We have searched different sites of insertion (homologies) and in our default modular design we have settled for far on a using the sites tried out by Prof Purton's lab (trnE2-psbH) for the driver (see picture below) and also a site which in literature is reported as producing high yields (TrnI-TrnA) for our gene of interest. Right?
MODULARITY AND COMPATIBILITY
- We design the system highly modular and compatible with Golden Gate so that it can be easily used and changed, it should work with any standard strain of Chlamydomonas. We are looking into possibility of maintaining our system modular by using GoldenBraid technique.
Well done if you got up to here! Have a look at our current design:
- First we have abandoned the idea of marking our genes with luciferase as that would not give us quick and easily detectable response. Fluorescent proteins should be more reliable although there are troubles with expressing them in Chlamydomonas chloroplasts - well, something for us to troubleshoot with trying our 18 possible FPs!
SAFETY - CAS9 REGULATION AND WAYS OF EXPRESSION
- We are very concerned with safety as our design may resemble a gene drive, we want to make sure we are definitely not making one. We were considering Lac regulatable promoters, light regulated ones and also Nac2. Nac2 is still a system we consider to try - we could switch off the expression of Cas9 when needed and regulate it in case it's toxic in higher amounts by concentration of thiamin. But it is actually in the UTR and regulated by a nuclear gene which regulates a photosynthetic gene. So we need to obtain a strain which has been designed to have these pathways independent, we'd need to optimise concentrations of thyamin and design more constructs.!
- We also considered another level of safety by introducing another gRNA which would make the Cas9 to cleave itself after homoplasmy has been achieved. However as NHEJ does not exist in chloroplasts, we would also need to supply the template. Still an option! But at the moment we plan to introduce Cas9 cassette either transiently or just into one copy of DNA, not homoplasmic (we plan to try both), let it drive the propagation of our genes of interest and then be lost when we remove the selection pressure of antibiotics. A semi-stable type of integration was decided as an alternative to transient because transient may not produce enough product and be list too quickly.
- We are also considering delivery of the preassembled complex of gRNA and Cas9 or its parts into chloroplast by biolistics, although it might be beyond our time-scope
- Because of safety our Cas9 and gRNA were initially separated either physically or one of them was outside the homology regions so that it won't be integrated. It was because we had only one insertion site and corresponding homologies. In the current design the Cas9 and gRNA are in one construct but gRNA is targeting another region on the chloroplast genome (so that it drives expression).
- Our polycistronic design of cassette might be simpler and produce shorter DNA fragments but maybe complicated for cell processing so we decided to put separate promoters for gRNA, antibiotic resistance and Cas9 fused with FP
ACTIVITY OF CAS9
- We had troubles with assessing that our Cas9 is active as one of the first steps. It would be very elegant to let it drive propagation of its own cassette with markers (or changing colours of algae already transformed with a fluorescent protein), making the strain less viable in field but the same in the lab (eg. by nutrient utilisation defficiency). The best solution would be restoring functionality of some mutant strain by the cassette. But we want to avoid a gene drive at all costs. We have also considered surveyor assay. So we are still looking for ideal solutions, probably by tracking Cas9 cuts by qPCR. (or do we have a solution?)
- Our Cas9 is now fused with the fluorescent protein. It means each molecule of the FP corresponds to a molecule of Cas9. And we can supply an interesting part into the registry
TYPES OF CAS9
- We were also considering different versions of Cas9 - from different bacteria, expressed from nucleus targeted to chloroplast (unluckily it is probably toxic in the cytoplasm), transiently or stably expressed, and for long we were considering using nickase (which makes just single-stranded breaks and so pair of gRNAs is used to reduce off target activity by around hundreds of folds). But then we learned the toxicity is more likely because of sucking RNAs rather than cutting off-target.
WORKING SYSTEM
- We considered prototyping experiments on isolated chloroplasts to speed them up but at the moment we plan to transform chloroplasts in the whole Chlamydomonas
INSERTION SITES
- This design also enables any researcher with a prepared cassette for insertion to work with our system just by changing the gRNA (20 nucleotides) to correspond to the desired site of insetrion. We have searched different sites of insertion (homologies) and in our default modular design we have settled for far on a using the sites tried out by Prof Purton's lab (trnE2-psbH) for the driver (see picture below) and also a site which in literature is reported as producing high yields (TrnI-TrnA) for our gene of interest. Right?
MODULARITY AND COMPATIBILITY
- We design the system highly modular and compatible with Golden Gate so that it can be easily used and changed, it should work with any standard strain of Chlamydomonas. We are looking into possibility of maintaining our system modular by using GoldenBraid technique.
Well done if you got up to here! Have a look at our current design:
- We are very concerned with safety as our design may resemble a gene drive, we want to make sure we are definitely not making one. We were considering Lac regulatable promoters, light regulated ones and also Nac2. Nac2 is still a system we consider to try - we could switch off the expression of Cas9 when needed and regulate it in case it's toxic in higher amounts by concentration of thiamin. But it is actually in the UTR and regulated by a nuclear gene which regulates a photosynthetic gene. So we need to obtain a strain which has been designed to have these pathways independent, we'd need to optimise concentrations of thyamin and design more constructs.!
- We also considered another level of safety by introducing another gRNA which would make the Cas9 to cleave itself after homoplasmy has been achieved. However as NHEJ does not exist in chloroplasts, we would also need to supply the template. Still an option! But at the moment we plan to introduce Cas9 cassette either transiently or just into one copy of DNA, not homoplasmic (we plan to try both), let it drive the propagation of our genes of interest and then be lost when we remove the selection pressure of antibiotics. A semi-stable type of integration was decided as an alternative to transient because transient may not produce enough product and be list too quickly.
- We are also considering delivery of the preassembled complex of gRNA and Cas9 or its parts into chloroplast by biolistics, although it might be beyond our time-scope
- Because of safety our Cas9 and gRNA were initially separated either physically or one of them was outside the homology regions so that it won't be integrated. It was because we had only one insertion site and corresponding homologies. In the current design the Cas9 and gRNA are in one construct but gRNA is targeting another region on the chloroplast genome (so that it drives expression).
- Our polycistronic design of cassette might be simpler and produce shorter DNA fragments but maybe complicated for cell processing so we decided to put separate promoters for gRNA, antibiotic resistance and Cas9 fused with FP
ACTIVITY OF CAS9
- We had troubles with assessing that our Cas9 is active as one of the first steps. It would be very elegant to let it drive propagation of its own cassette with markers (or changing colours of algae already transformed with a fluorescent protein), making the strain less viable in field but the same in the lab (eg. by nutrient utilisation defficiency). The best solution would be restoring functionality of some mutant strain by the cassette. But we want to avoid a gene drive at all costs. We have also considered surveyor assay. So we are still looking for ideal solutions, probably by tracking Cas9 cuts by qPCR. (or do we have a solution?)
- Our Cas9 is now fused with the fluorescent protein. It means each molecule of the FP corresponds to a molecule of Cas9. And we can supply an interesting part into the registry
TYPES OF CAS9
- We were also considering different versions of Cas9 - from different bacteria, expressed from nucleus targeted to chloroplast (unluckily it is probably toxic in the cytoplasm), transiently or stably expressed, and for long we were considering using nickase (which makes just single-stranded breaks and so pair of gRNAs is used to reduce off target activity by around hundreds of folds). But then we learned the toxicity is more likely because of sucking RNAs rather than cutting off-target.
WORKING SYSTEM
- We considered prototyping experiments on isolated chloroplasts to speed them up but at the moment we plan to transform chloroplasts in the whole Chlamydomonas
INSERTION SITES
- This design also enables any researcher with a prepared cassette for insertion to work with our system just by changing the gRNA (20 nucleotides) to correspond to the desired site of insetrion. We have searched different sites of insertion (homologies) and in our default modular design we have settled for far on a using the sites tried out by Prof Purton's lab (trnE2-psbH) for the driver (see picture below) and also a site which in literature is reported as producing high yields (TrnI-TrnA) for our gene of interest. Right?
MODULARITY AND COMPATIBILITY
- We design the system highly modular and compatible with Golden Gate so that it can be easily used and changed, it should work with any standard strain of Chlamydomonas. We are looking into possibility of maintaining our system modular by using GoldenBraid technique.
Well done if you got up to here! Have a look at our current design:
- We had troubles with assessing that our Cas9 is active as one of the first steps. It would be very elegant to let it drive propagation of its own cassette with markers (or changing colours of algae already transformed with a fluorescent protein), making the strain less viable in field but the same in the lab (eg. by nutrient utilisation defficiency). The best solution would be restoring functionality of some mutant strain by the cassette. But we want to avoid a gene drive at all costs. We have also considered surveyor assay. So we are still looking for ideal solutions, probably by tracking Cas9 cuts by qPCR. (or do we have a solution?)
- Our Cas9 is now fused with the fluorescent protein. It means each molecule of the FP corresponds to a molecule of Cas9. And we can supply an interesting part into the registry
TYPES OF CAS9
- We were also considering different versions of Cas9 - from different bacteria, expressed from nucleus targeted to chloroplast (unluckily it is probably toxic in the cytoplasm), transiently or stably expressed, and for long we were considering using nickase (which makes just single-stranded breaks and so pair of gRNAs is used to reduce off target activity by around hundreds of folds). But then we learned the toxicity is more likely because of sucking RNAs rather than cutting off-target.
WORKING SYSTEM
- We considered prototyping experiments on isolated chloroplasts to speed them up but at the moment we plan to transform chloroplasts in the whole Chlamydomonas
INSERTION SITES
- This design also enables any researcher with a prepared cassette for insertion to work with our system just by changing the gRNA (20 nucleotides) to correspond to the desired site of insetrion. We have searched different sites of insertion (homologies) and in our default modular design we have settled for far on a using the sites tried out by Prof Purton's lab (trnE2-psbH) for the driver (see picture below) and also a site which in literature is reported as producing high yields (TrnI-TrnA) for our gene of interest. Right?
MODULARITY AND COMPATIBILITY
- We design the system highly modular and compatible with Golden Gate so that it can be easily used and changed, it should work with any standard strain of Chlamydomonas. We are looking into possibility of maintaining our system modular by using GoldenBraid technique.
Well done if you got up to here! Have a look at our current design:
- We were also considering different versions of Cas9 - from different bacteria, expressed from nucleus targeted to chloroplast (unluckily it is probably toxic in the cytoplasm), transiently or stably expressed, and for long we were considering using nickase (which makes just single-stranded breaks and so pair of gRNAs is used to reduce off target activity by around hundreds of folds). But then we learned the toxicity is more likely because of sucking RNAs rather than cutting off-target.
WORKING SYSTEM
- We considered prototyping experiments on isolated chloroplasts to speed them up but at the moment we plan to transform chloroplasts in the whole Chlamydomonas
INSERTION SITES
- This design also enables any researcher with a prepared cassette for insertion to work with our system just by changing the gRNA (20 nucleotides) to correspond to the desired site of insetrion. We have searched different sites of insertion (homologies) and in our default modular design we have settled for far on a using the sites tried out by Prof Purton's lab (trnE2-psbH) for the driver (see picture below) and also a site which in literature is reported as producing high yields (TrnI-TrnA) for our gene of interest. Right?
MODULARITY AND COMPATIBILITY
- We design the system highly modular and compatible with Golden Gate so that it can be easily used and changed, it should work with any standard strain of Chlamydomonas. We are looking into possibility of maintaining our system modular by using GoldenBraid technique.
Well done if you got up to here! Have a look at our current design:
- We considered prototyping experiments on isolated chloroplasts to speed them up but at the moment we plan to transform chloroplasts in the whole Chlamydomonas
INSERTION SITES
- This design also enables any researcher with a prepared cassette for insertion to work with our system just by changing the gRNA (20 nucleotides) to correspond to the desired site of insetrion. We have searched different sites of insertion (homologies) and in our default modular design we have settled for far on a using the sites tried out by Prof Purton's lab (trnE2-psbH) for the driver (see picture below) and also a site which in literature is reported as producing high yields (TrnI-TrnA) for our gene of interest. Right?
MODULARITY AND COMPATIBILITY
- We design the system highly modular and compatible with Golden Gate so that it can be easily used and changed, it should work with any standard strain of Chlamydomonas. We are looking into possibility of maintaining our system modular by using GoldenBraid technique.
Well done if you got up to here! Have a look at our current design:
- This design also enables any researcher with a prepared cassette for insertion to work with our system just by changing the gRNA (20 nucleotides) to correspond to the desired site of insetrion. We have searched different sites of insertion (homologies) and in our default modular design we have settled for far on a using the sites tried out by Prof Purton's lab (trnE2-psbH) for the driver (see picture below) and also a site which in literature is reported as producing high yields (TrnI-TrnA) for our gene of interest. Right?
MODULARITY AND COMPATIBILITY
- We design the system highly modular and compatible with Golden Gate so that it can be easily used and changed, it should work with any standard strain of Chlamydomonas. We are looking into possibility of maintaining our system modular by using GoldenBraid technique.
Well done if you got up to here! Have a look at our current design:
- We design the system highly modular and compatible with Golden Gate so that it can be easily used and changed, it should work with any standard strain of Chlamydomonas. We are looking into possibility of maintaining our system modular by using GoldenBraid technique.
Well done if you got up to here! Have a look at our current design:
2nd August
The day of meetings
In the morning we have met Cambridge Consultants to report our progress and discuss the cell selection and flow cytometry use as well as some hardware tips.
In the afternoon we had meeting with advisors discussing our long and short term goals.
Later we followed with planning of immediate experiments:
- Testing many different fluorescent protein markers in Chlamydomonas chloroplasts
- Testing toxicity of Cas9 in the chloroplast (it has been found toxic (probably RNA sponge) in the cytoplasm and cyanobacteria)
- Testing double selection on two antibiotics and the interaction of antibiotics and their resistances + cotransfection with two constructs in general
- Plus we plan to put our parts in iGEM backbones soon after they come
Group meetings are also very important!

3th August
Designing and Ordering of Primers
4th August
Experimental preperation
11th August
Proper labwork, amplifying our mVenus fluorescent protein and the backbone to put it in Constructing primers for amplification of our DNA parts and especially Cas9 for future Gibson Assembly
16-17 August
18th August
UK meetup of iGEM teams!
19th August
Further amplification of our parts, planning of new design of our transformation vectors.
Biolistics again, this time successfully shot the mVenus ligated into a vector and the original vector with a different fluorescent protein as a control. Let’s wait to see if some genes got into our algae and produced any fluorescence!




