Difference between revisions of "Team:SUSTech Shenzhen/Notebook/Molecular"
| Line 170: | Line 170: | ||
==== Geco plasmid construction ==== | ==== Geco plasmid construction ==== | ||
| − | 1. Geco,Vector,Bla+2A Ligation with [[Team:SUSTech_Shenzhen/Notebook/Protocol# | + | 1. Geco,Vector,Bla+2A Ligation with [[Team:SUSTech_Shenzhen/Notebook/Protocol#T4_DNA_Ligase.28M0202.29 | '''T4 DNA Ligase''']](Vector:Insert=1:3). |
Vector (5.8 kbp): 5.5ng/μl | Vector (5.8 kbp): 5.5ng/μl | ||
Revision as of 09:49, 16 October 2016

Molecular Experiment
Notebook
Contents
- 1 2015
- 2 2016
2015
Dec.
13th
Geco plasmid construction
1. Polymerase Chain Reaction (PCR)
Use PBX-084 TRE-mCherry-2A-Bla plasmid as template to get blasticidin(Bla) resistance gene.
Primers: Age-Bla-S & Bla-E2A-AS
(without purification), 20μl, 30 cycles
| Volume/ul | |
|---|---|
| Buffer | 2 |
| Template | 0.5 |
| Polymerase(Taq) | 0.2 |
| dNTP | 1 |
| Primer | 0.5, 0.5 |
| ddH2O | 15.4 |
2. PCR
Use PBX-084 TRE-mCherry-2A-Bla plasmid as template to get 2A sequence.
Primers: E2A-S & E2A-Hind III-AS
(without purification), 20μl, 30 cycles
| Volume/ul | |
|---|---|
| Buffer | 2 |
| Template | 0.5 |
| Polymerase(Taq) | 0.2 |
| dNTP | 1 |
| Primer | 0.5, 0.5 |
| ddH2O | 15.4 |
3. PCR
Step 1: run 10 cycles without primer to let Bla and 2A link together.
| 98℃ | 10s | \ | |
| 55℃ | 5s | x10 cycles | |
| 72℃ | 30s | / |
Step 2:add primers to run 30 cycles.
Primers: AgeI-Bla-S 0.3μM & E2A-HindIII-AS 0.3μM
| 98℃ | 10s | \ | |
| 55℃ | 5s | x30 cycles | |
| 72℃ | 30s | / |
4. PCR product cycle purification by using E.Z.N.A.® Cycle Pure Kit.
15th
Geco plasmid construction
1. Digest the PCR purification product with AgeI and HindIII restriction endonuclease(RE).(37 ℃ 2 hours)
| Volume/ul | |
|---|---|
| PCR purification product | 8.4 (~1ug) |
| CutSmart buffer | 5 |
| AgeI | 1.5 |
| HindIII | 1.5 |
| ddH2O | 33.6 |
2. Digest the Geco plasmid with BglII restriction endonuclease. (50℃ 3 hours)
| Volume/ul | |
|---|---|
| Geco plasmid | 9.5 (~1ug) |
| 3.1 buffer(X10) | 5 |
| BglII | 1.5 |
| ddH2O | 34 |
3.Cycle pure the RE digestion product with the kit “xxxxxx”.
4. Digest the purification product with HindIII restriction endonuclease. (37 ℃ 3 hours)
| Volume/ul | |
|---|---|
| the purification product | 5 (~640ng) |
| CutSmart (X10) | 5 |
| HindIII | 1 |
| ddH2O | 39 |
5. Digest the vector plasmid with AgeI restriction endonuclease. (37 ℃ 3 hours)
| Volume/ul | |
|---|---|
| The vector plasmid | 10 (~2.5 ug) |
| CutSmart(X10) | 5 |
| AgeI | 1 |
| ddH2O | 34 |
6.Add 1.5 μl BclII restriction endonuclease. (50 ℃ 3 hours)
Note: BclI and BglII have the same sticky end.
7. Gel running and Gel extraction to get the insert and vector fragments. E.Z.N.A.® Gel Extraction Kit - Spin Protocol
16th
Geco plasmid construction
1. Geco,Vector,Bla+2A Ligation with T4 DNA Ligase(Vector:Insert=1:3).
Vector (5.8 kbp): 5.5ng/μl
Bla+2A (0.45kbp): 16.8ng/μl
Geco (1.25kbp): 5.6ng/μl
| Kit | Add (ul) | |
|---|---|---|
| 10X T4 DNA Ligase Buffer | 2ul | 2.5 |
| Vector DNA(5.8kb) | 0.02pmol | 12.7 |
| Bla+2A(0.45kb) | 0.06pmol | 1 |
| Geco(1.25kb) | 0.06pmol | 8.2 |
| ddH2O | 0 | |
| T4 DNA Ligase | 1ul | 1 |
| Total | 20ul | 25 |
Room temperature for 10 minutes.
2. Mix&Go hyper-efficient competent cells transformation.
- Quickly thaw: remove "Mix&Go" hyper-efficient competent cells from -80℃ freezer and water flow till 50% of the cells melt.
- Add DNA: DNA volume is less than 1/10 volume of competent cells. (blow and suck for mixing )
- Heat shock: heat shock at 42℃ water bath for 45s.
- Spread the plates: remove LB(Amp+) plates from 4℃refrigerator,and spread all the bacterial on the plates. Overnight culture at 37℃ incubator (12~16hours).
Vector+Bla 2A+Geco 3 μl + competent cells 30 μl
17th
Geco plasmid construction
1. Vector+Bla 2A+Geco transformation results:

2. Pick single clones and overnight culture.
Pick 5 single clones and use 6 ml Amp+ LB medium with 37℃ and 220 rpm shaker to overnight culture each clones.
18th
Geco plasmid construction
1. Extraction of plasmid with E.Z.N.A.® Plasmid DNA Mini Kit I Protocol.
| Geco plasmid | Conc. (ng/ul) | A260/280 |
|---|---|---|
| 1 | 94.2 | 1.92 |
| 2 | 79.7 | 1.96 |
| 3 | 110.6 | 1.90 |
| 4 | 150.3 | 1.87 |
| 5 | 149.7 | 1.88 |
2. Send 5 plasmids to sequence.
29th
Piezo plasmid construction
1. Piezo backbone(PBX-123 plasmid from Prof.Huang’s lab) transformation. Mix&Go method.
Piezo backbone plasmids 2μl + competent cells 20μl
30th
Piezo plasmid construction
1. Piezo backbone transformation results:

2. Pick single clones and overnight culture.
Pick 3 single clones and use 6 ml Amp+ LB medium with 37℃ and 220 rpm shaker to overnight culture each clones.
31th
Piezo plasmid construction
1. Extraction of plasmid with E.Z.N.A.® Plasmid DNA Mini Kit I Protocol.
| piezo | Conc. (ng/ul) | A260/280 |
|---|---|---|
| 1 | 143.4 | 1.87 |
| 2 | 283.1 | 1.87 |
| 3 | 186.8 | 1.87 |
2. Piezo GPA、pTRE-3G PCR.
Piezo: 310ng/μl GPA: 444bp pTRE-3G: 376bp
| Protocol | Final conc. | Add (ul) | |
|---|---|---|---|
| PrimeSTAR Max(2X) | 25ul | 1X | 25 |
| Primer1 | 10-15pmol | 0.2-0.3uM | 1.3 |
| Primer2 | 10-15pmol | 0.2-0.3uM | 1.3 |
| Template | <200ng | 0.7 | |
| ddH2O | 21.7 | ||
| Total | 50ul | 50 |
Program setting:
| 98℃ | 10s | \ |
| 55℃ | 15s | X 35 cycles |
| 72℃ | 30s | / |
3.Gel Running

4.PCR purification.
E.Z.N.A.® Cycle Pure Kit - Centrifugation Protocol
| Conc. (ng/ul) | A260/280 | |
|---|---|---|
| GPA | 122.7 | 1.85 |
| pTRE-3G | 102.5 | 1.84 |
2016
Jan.
21th
Piezo plasmid construction
1. Piezo plasmid construction design:

2. Piezo plasmid(original) RE digestion: 37℃ 2hours
| Volume/ul | |
|---|---|
| piezo DNA (576ng/ul) | 2 |
| AscI | 1 |
| NotI | 1 |
| 10X CutSmart buffer | 5 |
| ddH2O | 41 |
3. Backbone PBX-123 plasmid RE digestion: 37℃ 2hours
| Volume/ul | |
|---|---|
| pBX123 DNA (312ng/ul) | 3 |
| MfeI | 1 |
| PmeI | 1 |
| 10X CutSmart buffer | 5 |
| ddH2O | 40 |
4. Gel running and gel extraction.
E.Z.N.A.® Gel Extraction Kit - Spin Protocol
| Gel Extraction concentration (ng/ul) | |
|---|---|
| piezo (original) | 18.6 |
| pBX123 | 40.7 |
5. Gibson assembly:
| Gibson system (20ul) | Gibson Control system(10ul) | |
|---|---|---|
| piezo | 4 | 0 |
| pBX-123 | 2 | 1 |
| pTRE-3G | 0.2 | 0.1 |
| GpA | 0.2 | 0.1 |
| Gibson mix (X2) | 10 | 5 |
| ddH2O | 3.6 | 3.8 |
Incubate the mix for 1 hour at 50°C. ( Gibson Assembly® Master Mix – Assembly (E2611) Method)
6. Gibson product transformation:
[[Team:SUSTech_Shenzhen/Notebook/Protocol#Transformation_.26_Competent_cell_recovery | Transformation & Competent cell recovery]
5μl gibson product + 50μl competent cells
Spread plates and overnight cultrue.
22th
Piezo plasmid construction
- Pick single colonies:
- pick 1 colony from control group, pick 5 colonies from piezo group.
- Add into 6ml Amp+ LB, shake at 37℃ and 220 rpm.
- Make Amp LB and agar.
- make two 500ml Amp+ LB medium and one 500ml Amp+ LB agar.
- pour 29 plates.
- Sterilization.
- We put bunch of tips and tubes for sterilization.
Piezo plasmid construction
- Extract the plasmids from the single colonies (1 control group and 5 PIEZO group):
| Conc. (ng/ul) | |
|---|---|
| Control group | 389.6 |
| PIEZO group 1 | 343.5 |
| PIEZO group 2 | 376.3 |
| PIEZO group 3 | 371.6 |
| PIEZO group 4 | 388.5 |
| PIEZO group 5 | 245.2 |
- Gel electrophoresis of the six plasmids:
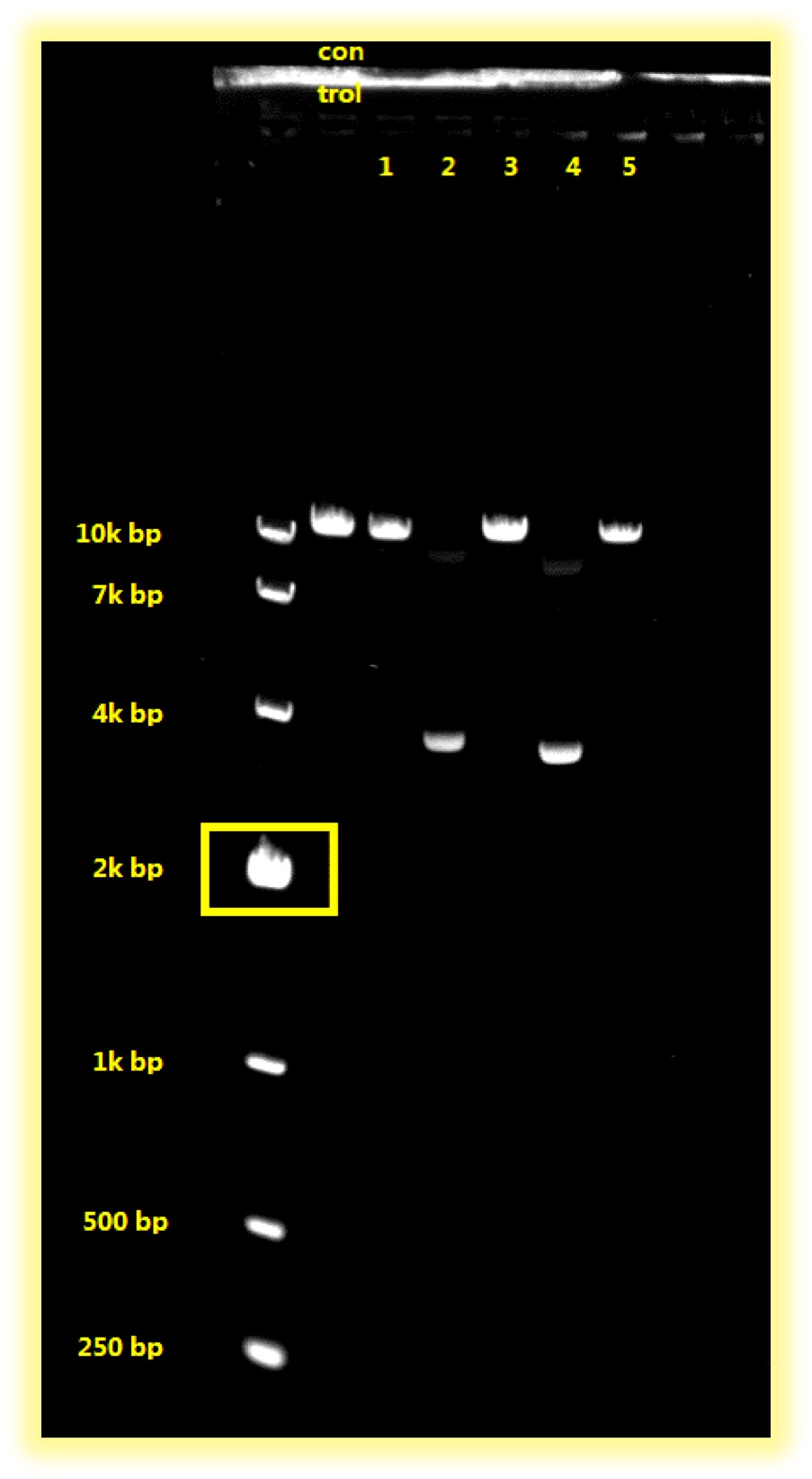

Sample 2 and Sample 4 have shown positive results, which should be tested further with PCR.
Materials sorting:
We sorted the material and put all of them into the lowermost layer of the -20℃ fridge.






25th
Piezo plasmid construction
- Sterilization:
- We put a bottle of ddH2O and bunch of tips and tubes for sterilization.
- PCR test for PIEZO group 2 and PIEZO group 4: Premix TaqTM (RR902A)
For each system
| Volume/ul | |
|---|---|
| Premix Taq | 10 |
| Sample | 0.5 |
| GpA primer1 | 1 |
| GpA primer2 | 1 |
| ddH2O | 7.5 |
Program setting:
| 95℃ | 5min | 95℃ | 30s | \ | 55℃ | 20s | x35 cycles | 72℃ | 40s | / |
3. Gel running:
The result showed that GpA had linked with backbone and piezo.
4. Pick up single colonies and colony PCR
As the result of gel electrophoresis of piezo Gibson group 2&4(yesterday,2016.1.24) missed bands of 4000, we picked up 20 single colonies to test by colony PCR. (use primers of GpA and PTRE).
The system of colony PCR (total 20 μl): Premix TaqTM (RR902A)
| Volume/ul | |
|---|---|
| 2X Taq mix | 10 |
| Template | 05 |
| Primer1 | 1 |
| Primer2 | 1 |
| ddH2O | 3 |
Program setting:
| 95℃ | 5min | ||
| 95℃ | 30s | \ | |
| 55℃ | 20s | x35 cycles | |
| 72℃ | 40s | / |
Finally, we got 40 groups for PCR colonies (20 for GpA and 20 for PTRE).
5. Gel electrophoresis of 40 groups after PCR colonies.
6.TRPC5 & TRPC6 &PBX-090 transformation.
We transformed these plasmids into competent cell and coated. After that, incubated at 37℃ for 16 hours.(5 plasmids onto 5 Amp plates)
26th
Piezo plasmid construction
1. Pick up single colonies
After TRPC5, TRPC6 and PBX-090 transformation, we found that there are no colony growing on PBX-090 plate. Later we knew that PBX-090 is Kanamycin resistant. We pick 1 colony from other 4 plates. Add into 5ml Amp LB, shake at 37℃.
2.PCR test 'Premix TaqTM (RR902A)
For yesterday PIEZO group 2 and PIEZO group 4(GPA verified):
| Volume/ul | |
|---|---|
| Premix Taq | 10 |
| Sample | 0.5 |
| GpA primer1 | 1 |
| GpA primer2 | 1 |
| ddH2O | 7.5 |
Program setting:
| 95℃ | 5min | ||
| 95℃ | 30s | \ | |
| 55℃ | 20s | x30 cycles | |
| 72℃ | 40s | / |
For yesterday PIEZO colony PCR promoter(pTRE) group 9,11,15(pTRE verified):
| Volume/ul | |
|---|---|
| Premix Taq | 10 |
| Sample | 0.5 |
| GpA primer1 | 1 |
| GpA primer2 | 1 |
| ddH2O | 7.5 |
Program setting:
| 95℃ | 5min | ||
| 95℃ | 30s | \ | |
| 55℃ | 20s | x30 cycles | |
| 72℃ | 40s | / |
3.Gel running of PCR product 346x376px
4.Streak plate of colony PCR9&11&15
We streaked 3 Amp plates using tips.
5.RE Digestion of PIEZO(original plasmid), PIEZO group 2,and PIEZO group 4 PIEZO Total 20μl
| Volume/ul | |
|---|---|
| 1ug PIEZO(original plasmid, conc. 576ng/ul) | 1.7 |
| BamHI | 0.2 |
| CutSmart buffer(X10) | 2 |
| ddH2O | 16.1 |
PIEZO group 2(1) Total 20μl
| Volume/ul | |
|---|---|
| 1ug PIEZO group2(original plasmid, conc. 376.3ng/ul) | 2.6 |
| PmeI | 0.2 |
| CutSmart buffer(X10) | 2 |
| ddH2O | 15.2 |
PIEZO group 2(2) Total 20μl
| Volume/ul | |
|---|---|
| 1ug PIEZO group2(original plasmid, conc. 376.3ng/ul) | 2.6 |
| PmeI | 0.2 |
| BamHI | 0.2 |
| CutSmart buffer(X10) | 2 |
| ddH2O | 15 |
PIEZO group 4(1) Total 20μl
| Volume/ul | |
|---|---|
| 1ug PIEZO group4(original plasmid, conc. 388.5ng/ul) | 2.6 |
| PmeI | 0.2 |
| CutSmart buffer(X10) | 2 |
| ddH2O | 15.2 |
PIEZO group 4(2) Total 20μl
| Volume/ul | |
|---|---|
| 1ug PIEZO group2(original plasmid, conc. 388.5ng/ul) | 2.6 |
| PmeI | 0.2 |
| BamHI | 0.2 |
| CutSmart buffer(X10) | 2 |
| ddH2O | 15 |
Gel running results: 366x367px
Since the results out to be blurred, we decide to run gel again tomorrow.
27th
Piezo plasmid construction
1.Plasmid purification E.Z.N.A.® Plasmid DNA Mini Kit I Protocol - Spin Protocol
Plasmid purification of TRPC5 and TRPC6 from the single colonies we picked yesterday
The concentration of the plasmids:
| Conc. (ng/ul) | |
|---|---|
| TRPC5 IRES-GFP | 461.9 |
| TRPC5 IRES-GFP | 425.1 |
| TRPC5 IRES-GFP | 542.7 |
| TRPC5 IRES-GFP | 866.8 |
2.Gel electrophoresis of Restriction Enzyme Digestion product: 369x336px
Something must have been wrong!
3.Pick single colonies After we strake plate of colony PCR9&11&15, we pick 1 colony from each plate. Add into 5ml Amp LB, shake at 37℃.
4. TRPC5 T5-GFP PCR Q5® High-Fidelity 2X Master Mix(M0492S)
| Volume/ul | |
|---|---|
| Q5 premix | 10 |
| Primer1 | 1 |
| Primer2 | 1 |
| TRPC5 T5-GFP | 0.8 |
| ddH2O | 7.2 |
| Total | 20 |
We make 2 groups of 50μL PCR solution.
5. PCR purification E.Z.N.A.® Cycle Pure Kit - Centrifugation Protocol
Accidentally make the centrifuge in step 8 30 seconds rather than 1 minute
Use 50μl ddH2O to elute DNA in the last process.
DNA concentration:
Group1: 21.8ng/μL
Group2: 31.6ng/μL
6. TRPC5 T5-GFP PCR(2nd) Q5® High-Fidelity 2X Master Mix(M0492S)
Because the DNA concentration in the last step was too low to conduct the following experiment, we did the TRPC5 T5-GFP PCR for the second time.
| Volume/ul | |
|---|---|
| Q5 premix | 25 |
| Primer1 | 2.5 |
| Primer2 | 2.5 |
| TRPC5 T5-GFP | 3 |
| ddH2O | 17 |
| Total | 50 |
We make 5 groups of 50μL PCR solution, and run 40 cycles.
28th
Piezo plasmid construction
1. Plasmid purification and maintenance breeding ① Plasmid purification of colony PCR9&11&15 from the single colonies we picked yesterday
Use 50μL ddH2O to elute DNA in the last process.
E.Z.N.A.® Plasmid DNA Mini Kit I Protocol - Spin Protocol
The concentration of the plasmids:
Colony PCR9 463.6ng/μl
Colony PCR11 556.1ng/μl
Colony PCR15 474.4ng/μl
② Maintenance breeding
Mix 500μL bacterial liquid with 500μL (volume fraction:50%)glycerol, and then shore at -80℃.
2. PCR purification E.Z.N.A.® Cycle Pure Kit - Centrifugation Protocol
PCR purification of TPRC5 T5-GFP(2nd)
Use 80 μl Tris-HCl to elute DNA in the last process.
Finally, the concentration of DNA:
Group 1 55.8ng/μl
Group 2 68.7ng/μl
Group 3 69.2ng/μl
Group 4 71.6ng/μl
3. Mycoplasma detection
We did mycoplasma detection for the supernatant from the cell culture (step 4. Cell freezing) we collected yesterday.
a. PCR the supernatant by the primers of mycoplasma DNA.
The system of PCR (total 50μL): Q5® High-Fidelity 2X Master Mix(M0492S)
| Volume/ul | |
|---|---|
| Q5 hot start | 25 |
| Primer1 | 2.5 |
| Primer2 | 2.5 |
| DNA | 3 |
| ddH2O | 17 |
Program setting:
| 95℃ | 30s | ||
| 98℃ | 20s | \ | |
| 50℃ | 10s | x35 cycles | |
| 72℃ | 100s | / | |
| 72℃ | 120s | ||
| 4℃ | ∞ |
b. Gel electrophoresis of PCR product.
264x301px Obviously, positive control turns out to be positive result, while negative control and the samples are negative results, so that our freezing cells are not polluted and we can store them into liquid nitrogen (big liquid nitrogen cell storage tank 3-4).
4.Restriction Enzyme Digestion(A)
| Volume/ul | |
|---|---|
| DNA (~50ng/uL)(actually, we use group 1&2 from step 2. today) | 80 |
| Age I-HF | 1 |
| 10X CutSmart buffer | 9 |
Digest the ends of TRPC5 for ligation.
Since we have the PBX123 with sticky ends of Age I and Bcl I, we should digest TRPC5 with Age I enzyme and Bcl I.
Firstly, we digest TRPC5 with Age I (activity:100%).The system (total 90μL):
Incubate at 37℃ for two hours.
Then, we digest TRPC5 with Bcl I (activity:75%).
We add Bcl I enzyme (1μL) into the final system of the first digestion. Incubate at 50℃ for six hours.
Digest the product of plasmid purification (step 1. today).
We digest the product of plasmid purification of pick up single colonies of streak plate of colony PCR group9&11&15 to test its linking.
The system (total 20μL):
| Volume/ul | |
|---|---|
| DNA(~50ng/ul) | 1 |
| Bam HI-HF | 10.2 |
| 10X CutSmart buffer | 2 |
| ddH2O | 16.8 |
Incubate at 37℃ for two hours.
5. Gel electrophoresis of the digestion of plasmid purification product of colonies PCR 9&11&15 264x495px
As we digest the plasmid with only one enzyme, we only get linear 15k bp DNA. Obviously, group9&15 are positive.
6. Restriction Enzyme Digestion(B) and gel electrophoresis
We decide to digest the product of plasmid purification of colony PCR group 9&15 with Bam-HI and Bgl I to ensure they are exactly correct.
The system (total 20μL):
Bam HI-HF 0.2μL(activity:100%)
Bgl I 0.2μL(activity:100%)
DNA(~500ng/μL) 1.25μL
10×NEBuffer 2μL(3.1 buffer)
ddH2O 16.35μL
Incubate at 37℃ for two hours.
Gel electrophoresis of the product of digestion.
7. Restriction Enzyme Digestion purification E.Z.N.A.® Cycle Pure Kit - Centrifugation Protocol
Use 30μL ddH2O to elute DNA in the last process.
TRPC5 T5-GFP 1 72.6ng/Μl
TRPC5 T5-GFP 2 71.3ng/μL
8. Restriction Enzyme Digestion E.Z.N.A.® Cycle Pure Kit - Centrifugation Protocol
We digest PBX-123 with Age I to get the sticky end which can connected with TPRC5.
The system (total 50μL):
Age I-HF 2μL
DNA(~300ng/μL) 20μL(actually, we use PBX group 3.)
10×NEBuffer 5μL(CutSmart buffer)
ddH2O 23μL
Incubate at 37℃ for two hours.
9. Ligation (TRPC5 and PBX-123)
The system (total 20μL):
10×T4 DNA buffer 2μL
Vector(PBX123) 10μL(43.26ng)
TRPC5 1μL(55.26ng)
ddH2O 6μL
T4 DNA ligase 1μL
10. Transformation:
We transform the plasmid which we get in the step 9. Ligation into 25μL competent cell.
We also help Dr. Huang (our mentor) transform 3 plasmids into competent cell (each 25μL).
Then, we coat and incubate at 37℃for 16 hours.
29 th
Piezo plasmid construction
- Restriction Enzyme Digestion(PBX-123, Age I& Bcl I)
We digested PBX-123 with Age I to get the sticky end which can connected with TPRC5.
Then we use Bcl1 to digest.
The system (total 50μL):
Bcl I 2μL
DNA(~300ng/μL) 20μL (actually, we use PBX group 3.)
10×NEBuffer 5μL(CutSmart buffer)
ddH2O 23μL
Incubate at 50℃ for two hours.
{kind=link}
The result is strange. The brightest band is larger than 10000.
Still, we extract the plasmid from the brightest band. Concentration:61.8ng/μl
Gel running of restriction enzyme digestion (PIEZO Gibson Ligation from colony PCR group 9&15, BamHI Bgl II) 270x467px
The undigested ones showed more bands while digested one only showed one band which is really strange.
We ran the gel for two times and still got the same results.
3. Colony PCR (5 PIEZO Gibson colony &2 PIEZO Gibson control colony&Negative control ddH2O, PCR for GpA& PTRE)
Total 20μl
2xTaq 10μl
Template 5μl
Primer 1 1μl
Primer 2 1μl
DdH2O 3μl
Primers: GpA primers/PTRE primers
From the results, GpA can still be seen in ddH2O. The result is so strange. We don’t know how to explain.
4.Colony PCR (12 TRPC5 Ligation colonies&dd H2O)
Total 20μl
2xTaq 10μl
Template 5μl
TRPC5 Primer 1 1μl
TRPC5 Primer 2 1μl
ddH2O 3μl
The PCR results showed no correct one. We can’t see TRPC5 band.
The rightmost one: Digested PBX123 backbone for TRPC5 is the gel extraction from step 1(Conc. 61.8ng/μl).The band should be around 8000bp. While the result is larger than 10000bp. This indicates that the extracted plasmid is not right. </blockquote> 5. Restriction enzyme digest(PBX123 backbone for PIEZO, PIEZO, both MfeI&BamHI&PmeI)
- PBX123 backbone for PIEZO
Total 50μl
PBX123(~300ng/μl) 35 μl
10x Buffer 5 μl
DdH2O 7 μl
MfeI 1μl
BamHI 1μl
PmeI 1μl
- PIEZO
Total 50μl
PBX123(~570ng/μl) 20μl
10x Buffer 5 μl
DdH2O 22 μl
MfeI 1μl
BamHI 1μl
PmeI 1μl
6. Pick up single colonies
Since the results from the Colony PCR of TRPC5 ligation was not very promising, we pick up 6 colony from the plate. Add into 5ml Amp LB, shake at 37℃.
The signal colonies are picked for the PCR process to make sure that Colony PCR it self have nothing to do with the failure.
30th
Piezo plasmid construction
1. Enzyme digestion of PBX123(Age I and Bcl I for TRPC5)
PBX123(~300ng/μL) 30μL
Age I 1μL
CutSmart 5μL
ddH2O 14μL
Incubate in 37℃ for a whole night.
Add in Bcl I at 11:00, and move to 50℃.
Bcl I 1μL
Incubate in 50℃ for a 4 hours.
2. Gel electrophoresis of PBX123(Mfe I, BamH I and Pme I for piezo), PBX123(Age I and Bcl I for TRPC5) and Piezo
The PBX123(MfeI BamHI PmeI for piezo) and Piezo(digested) on the other gel was cut off for gel extraction
The concentration of the extraction:
PBX-123 40.8ng/μL
Piezo 28.4ng/μL
3. Plasmid extraction E.Z.N.A.® Plasmid DNA Mini Kit I Protocol - Spin Protocol
Plasmid extraction from the single colonies we picked up yesterday.
The concentration of the plasmids:
TRPC5 ligation 1 378.2ng/μL
TRPC5 ligation 2 347.8ng/μL
TRPC5 ligation 3 343.3ng/μL
TRPC5 ligation 4 441.5ng/μL
TRPC5 ligation 5 438.0ng/μL
TRPC5 ligation 6 431.0ng/μL
4. TRPC5 PCR
We add in the primer of TRPC5 and try to make TRPC5 PCR from the ligation plasmid we extracted from the single colonies.
We also make two control groups, positive control group from the original TRPC5 T5-GFP plasmid we got, and negative control group from ddH2O.
rTaq premix 5μL
Templet 1μL
Primer1 0.5μL
Primer2 0.5μL
ddH2O 3μL
5. Gel electrophoresis of the PCR products and the gel extraction products.
Nothing, even with the positive control group from the original TRPC5 T5-GFP plasmid we got, have shown the right result (some brightness around 3000bp).
6. Gibson assembly of Piezo and PBX123
Gibson Assembly® Master Mix – Assembly (E2611)
| Gibson group (10ul) | Control group1 (10ul) | Control group2 (10ul) | Control group3 (10ul) | |
|---|---|---|---|---|
| Piezo (28.4ng/ul) | 2.5ul | |||
| pBX-123 (40.8ng/ul) | 1.5ul | 1.5ul | 1.5ul | 1.5ul |
| pTRE-3G (102.5ng/ul) | 0.13ul | 0.13ul | ||
| GpA (122.7ng/ul) | 0.11ul | 0.11ul | ||
| Gibson mix | 5ul | 5ul | 2.5ul | 5ul |
| ddH2O | 0.76ul | 3.26ul | 1ul | 1ul |
7. Enzyme digestion of PBX123(Age I and Bcl I for TRPC5)(2nd)
PBX123(~300ng/μL) 30μL
Bcl I 1μL
3.1 5μL
ddH2O 14μL
Incubate in 50℃ for a 4 hours.
Conduct DNA extraction with Cycle-Pure Kit, use 45μL ddH2O to elute the DNA.
The DNA concentration was 92.5ng/μL.
DNA extraction 45μL
Age I 1μL
CutSmart 5μL
Incubate in 37℃ overnight.
8. Transformation
We transform the plasmids which we get in Gibson assembly into 33μL competent cell(1 gibson system and 4 control systems).
We also transform PBX090 and PBX123 to get more plasmid for further use.
31th
Piezo plasmid construction
- Gel electrophoresis of digested PBX123(Age I and Bcl I for TRPC5)(2nd)251x437px
- Colony PCR
Before colony PCR, we remove the plate of PBX123 from incubator to cold room.
We pick up 20 single colonies from the plate of Gibson group, 2 colonies from both control group 1 and 2. There are no colonies on plates of control group 3 and PBX090.
The system of colony PCR(use primers of poly A and pTRE):
Primer1 0.5μL
Primer2 0.5μL
Bacterial liquid 5μL
rTaq premix 6μL
We use original PBX123 to substitute the Bacterial liquid in positive control group 1.
We use poly A or pTRE that we get from the original PBX123 by PCR process to substitute the Bacterial liquid in positive control group 2.
We use ddH2O to substitute the Bacterial liquid in negative control group.
| 95℃ | 30s | ||
| 98℃ | 10s | \ | |
| 50℃ | 10s | x30 cycles | |
| 72℃ | 100s | / | |
| 72℃ | 120s | ||
| 16℃ | ∞ |
3. Gel electrophoresis of colony PCR (step 2.)
Obviously, every Gibson group are negative.
4. Enzyme digestion of PBX133(Bcl I and Age I, Nhe I, Xba I )
We suspect the availability of our Bcl I enzyme, so we digest this new backbone with another tube of Bcl I, and set a group digested by our enzyme to test its availability.
The system of digestion (by Bcl I of HW) (total 100μL):
Template(~200ng/μL) 50μL
Bcl I 5μL
NEBuffer(3.1) 10μL
ddH2O 35μL
Incubate in 50℃ for 1 hour.
The system of digestion (by Bcl I of IGEM)(total 10μL):
Template(~200ng/μL) 2.5μL
Bcl I(IGEM) 0.25μL
NEBuffer(3.1) 1μL
ddH2O 6.25μL
Incubate in 50℃ for 3.5 hours.
Conduct DNA extraction with Cycle-Pure Kit, use 50μL ddH2O to elute the DNA.
The DNA concentration was ng/μL.
The system of digestion (by Age I, Nhe I, Xba I )(total 50μL):
DNA extraction 42μL
Age I 1μL
Nhe I 1μL
Xba I 1μL
NEBuffer(CutSmart) 5μL
Incubate in 37℃ overnight.
5. Gel electrophoresis of PBX133 digested by Bcl I(after incubating for 1 hour) (step 4.)
229x496pxFrom the result we can ensure our enzyme is available.
Feb.
01 st
Piezo plasmid construction
1. Gel running of 4 enzymes digestion
{kind=link}
{kind=link}
For verification For gel extraction
{kind=link}
It’s easy to observe small fragments which is about 100-200bp. This indicates that the PBX133 backbone was cut.
Digestion sites: Bcl I,Age I,Nhe I,Xba I
2. Gel Extraction E.Z.N.A.® Gel Extraction Kit - Spin Protocol
We use the rest digested product to do gel extraction of cut PBX133(After 4 enzymes digested) Concentration:39.7 ng/μl
3. Transformation of enzyme digested products
360x406px2.5μl PBX133 digested by 4 enzymes, 2.5μl PBX133 digested by BclI(iGEM), 1μl PBX133 original, 2.5μl PBX133 digested by BclI(HW) with 25μl competent cells each.
{kind=link}
14 th
TRPC5 plasmid construction
1. Enzyme digestion of TRPC5(T5-GFP and IRES-GPF)
| Volume/ul | |
|---|---|
| TRPC5(~400ng/ul) | 0.5 |
| Not I | 0.2 |
| 10X CutSmart buffer | 1 |
| ddH2O | 8.3 |
| Total | 10 |
Incubate in 37℃ for 3 hours.
2. TRPC5 PCR(T5-GFP and IRES-GPF)
| Volume/ul | |
|---|---|
| Template | 1 |
| Primer1 | 2.5 |
| Primer2 | 2.5 |
| Q5 hotstart 2X Master Mix | 25 |
| ddH2O | 19 |
40 cycles
3. Make petro dishes.
We make 10 LB K+ petro dishes,75 Amp+ petro dishes and 14 LB petro dishes while waiting.
4. Gel electrophoresis of TRPC5
The expected brightness (~3k bp) appeared with TRPC5 T5-GFP PCR.
But TRPC5 PCR purification group 4, which we did with the same process as TRPC5 T5-GFP PCR. Did not have the same brightness. So we decided to do another Gel electrophoresis process tomorrow.
5. PCR purification of TRPC5 PCR products (T5-GFP and IRES-GPF)
E.Z.N.A.® Cycle Pure Kit - Centrifugation Protocol
The concentration of the products (diluted with 40μL ddH2O):
TRPC5 T5-GFP 105.2ng/μL
TRPC5 IRES-GPF 38.8ng/μL
6. Enzyme digestion of TRPC5 T5-GFP PCR purification product
Since we have the expected brightness (~3k bp) appeared with TRPC5 T5-GFP PCR, we carry on with the experiment. And started the enzyme digestion. Because Bcl I only have 75% activity in CutSmart buffer. So we decided to leave it overnight.
| Volume/ul | |
|---|---|
| TRPC5 T5 GFP(~102ng/ul) | 30 |
| Bcl I | 2 |
| CutSmart | 5 |
| ddH2O | 13 |
| Total | 50 |
Incubate in 50℃ for overnight.
15 th
TRPC5 plasmid construction
1. Enzyme digestion of TRPC5(T5-GFP-Age 1)
Add 2 μL Age 1 into the system of last-night enzyme digestion.
Incubate in 37℃ for 2 hours.
2. Enzyme digested product purification of TRPC5
E.Z.N.A.® Cycle Pure Kit - Centrifugation Protocol
The concentration of the products (diluted with 30μL ddH2O):
TRPC5 T5-GFP 58.8 ng/μl
3. Gel electrophoresis of used TRPC5
As we can see, the expected brightness (~3k bp) also appeared. It means that our TRPC5 PCR purification product is no problem, while the other four T5 products which were did before all have problem. So the four can’t be used, we will go deep by using the new TRPC5 PCR purification product.
4. Ligation of TRPC5 and PBX133 backbone T4 DNA Ligase(M0202)
| Volume/ul | |
|---|---|
| 10* T4 DNA Ligase Buffer | 2 |
| Vector DNA | 3 |
| Insert DNA | 2 |
| T4 DNA Ligase | 1 |
| ddH2O | 12 |
| Total | 20 |
Gently mix and let it in room temperature for 20 minutes.
Heat inactive at 65℃ for 10 minutes and put it in -20℃ for 4 hours.
5. Transformation of TRPC5 plasmid
| Competent cells/ul | DNA/ul | |
|---|---|---|
| TRPC5-pBX133 Ligation 1 | 33 | 5 |
| TRPC5-pBX133 Ligation 2 | 33 | 5 |
| pBX133 backbone | 33 | 5 |
Results :
{kind=link}
{kind=link}
TRPC5-PBX133 Ligation 2 TRPC5-PBX133 Ligation 1
{kind=link}
PBX133 backbone
The results are not so good. There are many kinds of passible reasons which we will go to confirm one by one tomorrow.
16
TRPC5 plasmid construction =
- Learn to design primer from Prof. Huang.
- Make new Amp+ agar plate.
- Spread 12μl 50mg/ml Amp+ (sodium salt) to the plate made in Feb.24
- Put the plate to 25℃ for 3 hours
- Examination of experiment and propagation of 090
Amp+ New plate(2016.2.16):
| Competent cells/ul | DNA/ul | |
|---|---|---|
| TRPC5-pBX133 Ligation 1 | 25 | 5 |
| pBX133 backbone 1 | 25 | 5 |
| TRPC5-pBX133 Ligation 2 | 25 | 1 |
| pBX133 backbone 2 | 25 | 1 |
| Control | 25 | 0 |
Amp+ Old plate(2016.2.14):
| Competent cells/ul | DNA/ul | |
|---|---|---|
| pBX133 backbone 1 | 25 | 5 |
| TRPC5-pBX133 Ligation 2 | 25 | 1 |
| pBX133 backbone 2 | 25 | 1 |
| Control | 25 | 0 |
Amp+ Former plate(2015):
| Competent cells/ul | DNA/ul | |
|---|---|---|
| TRPC5-pBX133 Ligation 1 | 25 | 5 |
K+ plate(2016.2.14):
| Competent cells/ul | DNA/ul | |
|---|---|---|
| 090 | 25 | 5 |
| Control | 25 | 0 |
Results:
Amp+ New plate(2016.2.16):
{kind=link}
{kind=link}
TRPC5-PBX133 Ligation 1 TRPC5-PBX133 Ligation 2
{kind=link}
{kind=link}
PBX133 backbone 1 PBX133 backbone 2
{kind=link}
Control
Amp+ Old plate(2016.2.14):
{kind=link}
{kind=link}
TRPC5-PBX133 Ligation 1 PBX133 backbone 1
{kind=link}
{kind=link}
PBX133 backbone 2 Control
Amp+ Former plate(2015):
{kind=link}
TRPC5-PBX133 Ligation 1
K+ plate(2016.2.14):
{kind=link}
{kind=link}
Control 090
Discussion:
Compare new and old plate, it comes out that some is wrong with the plate for its antibiotic function.
We thinks there may be two reason for this:
1.the antibiotics is inactived.
2.there is some distribution problem in the production of plate.
3.there is some concentration issue with the antibiotic.
There is also some problem with the ligation of TPRC6 and PBX133 backbone.
17 th
TRPC5 plasmid construction
- To check whether Ampicillin and Kanamycin are effective
- Add 1 μl Ampicillin solution to each LB agar plate and spread.Add 1 μl Kanamycin solution to each LB agar plate and spread.
- 2 Let them stay for 3 hours.
- 3 Add 25 μl competent cell to each plate. Incubate for 16 hours in 37 ℃
- Result:There is no any colony in each plate. So Ampicillin and Kanamycin are effective.
- TRPC5 primer design
{kind=link}
3. Make Amp+ and K+ LB agar plates.
We made 2*500ml LB agar. Add 500μl 1000x Amp+ in one and add 500 μl 1000x K+ in the other. We poured 15 K+ plates and 13 Amp+ plates.
18 th
TRPC5 plasmid construction
- Modify the previous designed primers,based on following defects
- Protection bases are too short,should be 4~6 bases.
- The temperature of matching pieces is too low,should be 55~65℃,and the Tm of primer should be 75~85℃.
- The number of bases between start codon and stop codon of final ligation product should be the fold of 3.
- Modified primers:
J23100-sfgfp-pBX123-piezo-primer
primer1:5'-AGAC ACCGGT TCTAGA CTAGAGTCGCGGCCGCTTTACTTGTA- 3'
AgeI XbaI Tm 62.6
primer2:5'-TACG GGATCC TTA T GGCGCGCC TTGATATCGAGCTCTTGACGGCTAGC- 3'
BamHI AscI Tm 60.9 J23100-sfgfp-pBX123-TRPC5-primer
primer1: 5'-AGAC ACCGGT GAATTC CTAGAGTCGCGGCCGCTTTACTTGTA- 3'
AgeI EcoRI Tm 62.6
primer2: 5'-TACG GGATCC TTA T GCGGCCGC TTGATATCGAGCTCTTGACGGCTAGC- 3'
BamHI NotI Tm 60.9
2. Amplify PBX-123 backbone
Backbone 1μL+competent cell 33μL (3 tubes)
Results:All 3 culture dishes don’t grow colony.
Discussion:Maybe the concentration of PBX-123 backbone is too low,so we increase the concentration.(2016.02.19)
3. Enzyme digestion of PBX-123 backbone(previous extracted PBX-123) 2 tubes,each 3μL
| NotI/ul | 1 |
| CutSmart/ul | 5 |
| pBX123 /ul | 3 |
| ddH2O | 41 |
| Total/ul | 50 |
Purification of PBX-123(30μL system,2 tubes)
Results:
①13.7ng/μL A260/280: 1.6
②20.6ng/μL A260/280: 1.4
4. Add Klenow Fragment to create blunt ends(2 tubes)
Klenow Fragment: 5units/μL
DNA Polymerase I, Large (Klenow) Fragment(M0210S)
Protocol(combine the NEB and Chinese protocol):
①
| Total | 40 | Final |
|---|---|---|
| 10X NEBuffer2 | 4 | |
| dNTP(2.5mM) | 0.6 | 33uM |
| pBX123 | 27.5 | 20~30ul(0.2~8ug) |
| Klenow Fragment | 0.1 | 0.4~1.6ul (2~8unit) 1 unit per ugDNA |
| Control | 7.8 |
②Incubate 15min at 25℃
③Incubate 30min at 75℃
5. Gel electrophoresis of 2 tubes blunt-ended PBX-123
Marker: DL10000
Results: Both 2 bands are near 10000bp and at the same site,which means the two bands are both blunt ends or sticky ends,we need to confirm after extracting the plasmid.
(We forgot to take a picture of the gel)
19 th
TRPC5 plasmid construction
1. Gel cutting and extraction of PBX-123 Klenow Fragment products
(30μL system)
Concentration:7.6ng/μL, A260/280: 1.55
2. Ligation of PBX-123 blunt end T4 DNA Ligase(M0202)
| Volume/ul | |
|---|---|
| Total | 30 |
| 10X Buffer | 3 |
| DNA | 25 |
| T4 DNA Ligase | 2 |
Amplify PBX-123 backbone,transform,select single colony and shake colony(4 tubes)
Results:
| PBX-123/ μl | Competent cell/ μl | Colonies |
| 0 | 25 | 0 |
| 5 | 25 | 10+ |
| 15 | 25 | 2 |
| 25 | 25 | 1 |
333x249px264x302px Fig.1 PBX-123 0 μl control Fig.2 PBX-123 5 μl
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Fig.3 PBX-123 15 μl Fig.4 PBX-123 25 μl
Discussion:
The number of colonies decreases as the concentration of PBX-123
backbone increases,which is strange,so we need to do a gel running after
extracting the plasmid to confirm whether the PBX-123 has problem.
4. Ligation product transform,select single colony and shake colony (1 tubes)
Results: only one culture dish grows 1 colony
{kind=link}
PBX-123 Not I-cut ligation 3
---
20 th
TRPC5 plasmid construction
1. PBX123 and PBX123(NotI-cut ligation) plasmid extraction
AxyPrep Plasmid Miniprep Spin Protocol
Final volume: 70 ul each tube
| PBX-123 NotI-cut | 249.3ng/ul |
| PBX-123 plasmid1 | 299.1ng/ul |
| PBX-123 plasmid2 | 213.6ng/ul |
| PBX-123 plasmid3 | 252.9ng/ul |
| PBX-123 plasmid4 | 360.3ng/ul |
2. Breed preservation :
500ul bacterial : 500ul glycerol each tube
| PBX-123 NotI-cut | 1 ml | -8℃ 107 |
| PBX-123 plasmid1 | 1 ml | -8℃ 107 |
| PBX-123 plasmid2 | 1 ml | -8℃ 107 |
| PBX-123 plasmid3 | 1 ml | -8℃ 107 |
| PBX-123 plasmid4 | 1 ml | -8℃ 107 |
3. NotI digest and transform (2 tubes)
| NotI/ul | 1 |
| CutSmart/ul | 5 |
| pBX-123/ul | 3 |
| ddH20/ul | 41 |
| Total/ul | 50 |
Purification: 40 ul system
Results: ① 18.1 ng/μL A260/280: 1.64
Transform 3 plates (Amp+)
4. He Yuhao repeats PBX123 NotI-cut:
NotI digest: 2 tubes
| NotI/ul | 1 |
| CutSmart/ul | 5 |
| pBX-123/ul | 3 |
| ddH20/ul | 41 |
| Total/ul | 50 |
Purification: 30 ul system each
Results: ① 25 ng/μL A260/280: 1.70
② 23 ng/μL A260/280: 1.66
21 th
TRPC5 plasmid construction
1.Select single colony
All 3 culture dishes grow many colonies,but we still need to do colony
PCR to confirm they are blunt-ended.Ling Shaohua has designed the primer
for colony PCR,we need to ask Pro.Huang whether it’s right.
So we select 10 colonies,each with 10ul Amp+ LB broth and store at cold
room.When the primer is verified and synthesized,we will do colony PCR
and extract plasmid from those colonies which are confirmed to be right.
{kind=link}
{kind=link}
PBX-123 Not I cut 2.20 ① PBX-123 Not I cut 2.20 ②
{kind=link}
PBX-123 Not I cut 2.20 ③
22 th
TRPC5 plasmid construction
1.Enzyme double digestion of HindIII-HF,Not I-HF. 37 ℃, 3hrs
| Volume/ul | |
|---|---|
| pBX-123 Not I-cut plasmid(~249ng/ul) | 4 |
| CutSmart buffer(10X) | 5 |
| HindIII-HF | 1 |
| NotI-HF | 1 |
| ddH2O | 39 |
| Volume/ul | |
|---|---|
| pBX-123 Not I-cut plasmid(~252ng/ul) | 4 |
| CutSmart buffer(10X) | 5 |
| HindIII-HF | 1 |
| NotI-HF | 1 |
| ddH2O | 39 |
567x436px Fig.1 PBX-123 plasmid pattern
{kind=link}
2.Gel running, proof:
318x606px This shows we have successfully delete the Not-I RE site.
{kind=link}
24 th
TRPC5 plasmid construction
1. TRPC5 enzyme digestion
We cut the TRPC5 PCR gel extraction from day 2.16 to give it a shot.
Only 23μL DNA left inside the tube.
| Volume/ul | |
|---|---|
| DNA | 23 |
| CutSmart buffer(10X) | 5 |
| BclI | 1 |
| ddH2O | 39 |
| Total | 50 |
2. Transformation:
We transform PBX133 PBX090 and Piezo to propagate them for further use.
We use the micropipette to inhale and blow the liquid inside Piezo for the plasmid sample, and add it to 33μL competent cells.
We add 2.5μL PBX133 and 2.5μL PBX090 to 33μL competent cells.
We coat and incubate at 37℃for 16 hours.
April
13 th~24 th
087&NFAT Plasmid Construction
Restriction Enzyme Digestion for Verification
{kind=link}
Gel Extraction
Sequencing Result:
Correct
04.24~05.06 TRPC5 Plasmid Construction
06.15~06.30 Restriction Enzyme,Ligase and Transformation Efficiency Test sfgfp PCR and Gel Extraction
{kind=link}
Transformation Result
{kind=link}
{kind=link}
July
0 1 st
Efficiency Tests
Transformation efficiency (3 groups)
| Competent cell/ul | sfGFP plasmid/ng | sfGFP plasmid/ul |
|---|---|---|
| 33 | 45.88 | 0.3 |
Enzyme digestion efficiency (3 groups)
| Competent cell/ul | sfGFP digested/ng | sfGFP digested/ul |
|---|---|---|
| 33 | 45.88 | 1.9 |
1>Prepare digestion system:
| Amount/ ul | |
|---|---|
| DNA | 2.4 |
| AflII | 1 |
| EcoRI | 1 |
| CutSmart(NEB) | 2.5 |
| ddH2O | 18.1 |
| Total | 25 |
Incubate at 37℃,6hrs.
Ligation efficiency (3 groups)
| Competent cell/ul | sfGFP plasmid ligated/ng | sfGFP plasmid ligated/ul |
|---|---|---|
| 33 | 41.67 | 3.3 |
1> Prepare ligation system T4 DNA Ligase(M0202)
| Vector | 0.02pmol |
| Insert | 0.08pmol |
| T4 ligase | 1ul |
| T4 ligase buffer 10X | 2ul |
| ddH2O | to 20ul |
| Total | 20ul |
Incubate at room temperature for 20mins
Transformation & Competent cell recovery
1. Get competent cell (DH5α) from -80℃,lay on ice for 15mins till melted.
2. Add plasmids according to the table above, gently blow to mix well.
3. Incubate on ice for 30mins.
4. Incubate in 42℃ water bath for 90s.
5. Lay on ice for 5mins.
6. Add 300μl LB, 37℃, 220rpm for 20mins.
7. Add 300μl Amp+ LB, 37℃, 220rpm for 20mins.
8. Centrifuge at 5000rpm, 1min. Drop 400μl suspension.
9. Mix the left 200μl gently, spread plate.
NOTE:
- For ligation products, subpackage of competent cell may decrease the final efficiency.
- During transformation, after heat shock, competent cell should be spread on preheated agar plate (37℃) to reach a higher transformation efficiency.
02 nd
Efficiency tests results:
| Green colony number | Non-green colony number | |
|---|---|---|
| Transformation 1 | >1000 | 24 |
| Transformation 2 | >1000 | 32 |
| Transformation 3 | >1000 | 21 |
| Digestion1 | 0 | 382 |
| Digestion2 | 1 | 370 |
| Digestion3 | 0 | 400 |
| Ligation1 | 0 | 300 |
| Ligation2 | 4 | 302 |
| Ligation3 | 0 | 267 |
NOTE:
Too many non-target bacterial colonies(non-green).
Efficiency tests failed.
Non-green colony test (I)
Pick 1 non-green colony on each plate.
Mix with 20μl ddH2O
Get 10μl add into 5ml Amp+ LB to incubate at 37℃, 220rpm for <16hrs.
Get 10μl to do colony PCR:
1> Primer
Forward -- AACGTATAAGCTTTAGGCGTGTACG
Reverse -- TTGTGCACCAGTCATAGCCGAATAG
2> Polymerase Chain Reaction (PCR)system</ul>
Q5® High-Fidelity 2X Master Mix(M0492S)
| Amount/ul | |
|---|---|
| Q5 Master Mix 2X | 12.5 |
| Colony mixture | 10 |
| PrimerF | 1 |
| PrimerR | 1 |
| ddH2O | 0.5 |
| Total | 25ul |
3> PCR setting
| STEP | TEMP | TIME |
|---|---|---|
| Initial Denaturation | 98℃ | 30s |
| 30 Cycles | 98℃ | 10s |
| 30 Cycles | 50-72℃ | 20s |
| 30 Cycles | 72℃ | 20s |
| Final Extension | 72℃ | 2mins |
| Hold | 10℃ | ∞ |
4> Gel electrophoresis
{kind=link}
03 th
Non-green colony test (II)
1. Use E.Z.N.A.TM Plasmid Mini Kit(Omega D6942-02) to purified plasmids.
According to spin protocol.
Test the concentration ( Eppendorf BioSpectrometer)
Use another pairs of primers to do PCR
Primer Forward -- CTAGAGTCGCGGCCGCTTTACTT
Primer Reverse -- TCGAGCTCTTGACAGCTAGCTCA
4.Double-enzyme digestion
| Non-green plasmid | 200ng |
| AflII | 1ul |
| EcoRI | 1ul |
| CutSmart(NEB) | 2ul |
| ddH2O | to 20ul |
| Total | 20ul |
5. Gel electrophoresis
04 th
Non-green colony test (III)
1.Enzyme digestion
| DNA | 200ng |
| ScaI | 1ul |
| CutSmart(NEB) | 2ul |
| ddH2O | to 10ul |
| Total | 10ul |
2. Gel electrophoresis
{kind=link}
06 th
1.Enzyme digestion
| Non-green plasmid | 200ng |
| ScaI/XbaI/XhoI | 1ul |
| CutSmart(NEB) | 2ul |
| ddH2O | to 20ul |
| Total | 20ul |
2. Gel electrophoresis
07 th-10 th
New plasmids design:
- TRPC5-Loxp
- Sfgfp-pBX123
- pBX097-neoloxp
11 th
TRPC5-Loxp Plasmid Construction(I)
1. TRPC5-R plasmid enzyme digestion
| TRPC5-R | 2ug |
| DraIII | 1ul |
| CutSmart(NEB) | 2ul |
| ddH2O | to 20ul |
| Total | 20ul |
2. Gel electrophoresis for gel extraction
{kind=link}
NOTE: Wrong length and faint bands
DraIII digestion site:
{kind=link}
12 th
TRPC5-Loxp Plasmid Construction(II)
1.TRPC5-L and TRPC5-R PCR
Primer F -- TTGGCAAAGAATTCCTCGAGG
Primer R -- GCCGATCATGCTGATATTGAGT
2.Gel electrophoresis for gel extraction
3.Gel Extraction (According to Gel Extraction protocol )
4.TRPC5-L plasmid re-synthesis
13 th
pBX097-neoloxp construction(I)
Neoloxp PCR from pBX123 plasmid
Gel extraction
Test product’s concentration
4.Double-enzyme digestion and pBX097 plasmid enzyme digestion
| pBX097/neoLoxP | 3ug |
| MfeI | 1ul |
| BamHI | 1ul |
| CutSmart(NEB) | 5ul |
| ddH2O | to 50ul |
| Total | 50ul |
5.Gel electrophoresis
{kind=link}
16 th - 21 th
pBX097-neoloxp construction(II)
1.Ligate the pBX097 and neoloxp with T4 DNA ligase and Quick Ligase (NEB)
2.DNA transformation
Result: None colony
TRPC5-Loxp Plasmid Construction(III)
TRPC5-R PCR product double-enzyme digestion (AflII; KpnI) overnight
TRPC5-L synthesis products transformation
TRPC5-L plasmid purification
TRPC5-L plasmid enzyme digestion (AflII; KpnI) overnight
Ligation of TRPC5 left and right (T4 DNA ligase)
Spread plate, incubate at 37℃ for <16hrs
Pick single colony, incubate in 15ml Amp+ LB for 16hrs at 37℃
Separate the culture into 5ml(to prove); 9.5ml(for endofree); 0.5ml (for
preservation in -80℃)
Single-enzyme digestion (PvuI; BamHI) and gel electrophoresis
Correct
pBX123-sfGFP construction
- sfGFP PCR
- Pbx123 double-enzyme digestion
3. Double-enzyme digestion ( AgeI; BamHI ) of sfGFP Overnight
4.Gel Extraction and Cycle pure
5. Ligation of sfGFP and pBX123 backbone
6. Spread plate incubate at 37℃, <16hrs
7. Pick 8 colonies incubated in 5ml Amp+ LB each, at 37℃ 220rpm for <16hrs
8. Plasmid purification
E.Z.N.A.® Plasmid DNA Mini Kit I Protocol - Spin Protocol
9. Double enzyme digestion for proof (AflII; EcoRI)
10.Gel electrophoresis
22 th - 23 th
pBX097-neoloxp construction(II)
1.Ligate the pBX097 and neoloxp with T4 DNA ligase and Quick Ligase (NEB)
2.DNA transformation
Result: 7 colonies
3. Pick colonies to 5ml Amp+ LB, incubate at 37℃, 220rpm for <16hrs.
4. Plasmid extraction with Plasmid Mini Kit (Omega)
5.Single-enzyme digestion
| pBX097/neoLoxP | 200ng |
| AflIII/ScaI | 1ul |
| CutSmart(NEB) | 2ul |
| ddH2O | to 20ul |
| Total | 20ul |
Incubate at 37℃, overnight.
24 th - 25 th
pBX097-neoloxp construction(II)
1.Gel electrophoresis
Result: pBX097-neoloxp successfully constructed.
26 th - 30 th
TRPC5-Loxp Plasmid Construction(IV)
1.TRPC5-Loxp endofree plasmid purification.
2.pBX097-neoloxp and TRPC5 sequencing
Primer for TRPC5---GCCGATCATGCTGATATTGAGT
Primer for pBX097-neoloxp---GGCAACGTGCTGGTTATTGTGC
Aug.
01 st - 02 nd
pBX123-sfGFP Efficiency test
| pBX123-sfGFP Efficiency Test(AflII, EcoRI) | Transformation |
| pBX123-sfGFP Efficiency Test(AflII, EcoRI) | Enzyme digestion |
| pBX123-sfGFP Efficiency Test(AflII, EcoRI) | Ligation |
Repeat the experiments steps mentioned in 7.1
03 rd - 04 th
pBX123-sfGFP enzymes’ digestion sites ligation ability
{kind=link}
| Groups | Combination |
|---|---|
| 1 | AgeI+BamHI |
| 2 | AgeI+AflI |
| 3 | EcoRI+BamHI |
| 4 | EcoRI+AflI |
1> Prepare digestion system, incubate overnight
2> Gel extraction to get different fragments
3> Ligation of T4 DNA ligase/Quick ligase
4> Transformation
Results:
| Groups | Colony number |
|---|---|
| AgeI+BamHI | 0 |
| AgeI+AflI | 0 |
| EcoRI+BamHI | non-green |
| EcoRI+AflI | non-green |
05 th
TRPC5-loxp sequencing results:
| TRPC5-(1) | 1 mismatch |
| TRPC5-(2) | Correct |
| TRPC5-(3) | 1 Failed |
| TRPC5-(4) | 1 Correct |
pBX097-neoloxp sequencing results:
| 097-neoLoxP-(1) | Correct |
| 097-neoLoxP-(2) | Correct |
| 097-neoLoxP-(3) | 3 mismatches |
06 th - 07 th
Non-green colony PCR
{kind=link}
09 th
Change NFAT-YFP to NFAT-EGFP(I)
Double enzyme digestion (AgeI, BamHI) pBX123 to get EGFP fragment
Double enzyme digestion (AgeI, BamHI) NFAT-YFP to get backbone Overnight
10 th
Change NFAT-YFP to NFAT-EGFP(II)
Gel extraction for EGFP and NFAT backbone
Ligation by T4 DNA ligase
Transformation
11 th - 13 th
Change NFAT-YFP to NFAT-EGFP(II)
- Pick single colony to culture in 15ml Amp+ LB for 16hrs
- Plasmid mini kit to get purified plasmid
- Enzyme digestion to prove
- Send to sequencing
22 th
TRPC5 Random mutagenesis(I)
| TRPC5 Random mutagenesis | Megaprimer PCR |
| TRPC5 Random mutagenesis | Gibson |
| TRPC5 Random mutagenesis | Enzyme Digestion |
Megaprimer PCR
1> Pre-experiments: T5 exonuclease digestion level according to time.
a. Use PrimerStar to do PCR of TRPC5 mutation region (~640bp)
b. Gel extraction to get TRPC5 mutation region
c. Set a series gradients of T5 digestion time: 0, 5, 10, 20mins
| T5 exonuclease digestion time | groups |
|---|---|
| 5 mins | 1,2 |
| 10 mins | 3,4 |
| 20 mins | 5,6 |
d. EDTA preparation
To get EDTA solution with final concentration of 11mM (Planing to add 1μl solution into
50μl to stop reaction). We should get 540mM stock solution.
- Weigh 1.08g EDTA powder add into 5ml ddH2O
- Use magnetic stirring bar to help dissolve
- Add NaOH powder to adjust solution’s PH until dissolved completely
- Use 0.22μm filter to filtrate the solution
- store in -20℃
- Use 0.22μm filter to filtrate the solution
- Add NaOH powder to adjust solution’s PH until dissolved completely
- Use magnetic stirring bar to help dissolve
Note: To keep T5 exonuclease digestion time as precise as possible, according to its protocol, we use EDTA to stop the digestion. e. T5 digestion (according to T5 exonuclease protocol)
| T5 exonuclease | 3.4ul |
| NEB4.0 Buffer | 5ul |
| TRPC5 region | 3.4ug |
| ddH2O | to 50ul |
| Total | 50ul |
f. Take group①②③for gel extraction; ④⑤⑥for cycle pure. 562x245px
{kind=link}
23 th
Megaprimer PCR 1> Send ④⑤⑥ to sequence Primer F:ATCCGAATTCCCCTCCAAATC Primer R:TATGTTAAGTTCCCAGCCCAG 2> Megaprimer PCR:
- Use Q5® High-Fidelity 2X Master Mix(M0492S), System 50μl, Tm=65℃
- Cycle pure to get purified PCR products.
- Use DpnI to digest plasmids that come from bacteria (methylated sites)
- Cycle pure to get purified PCR products.
318x146px **** Transformation of digested products.
{kind=link}
24 th
Megaprimer PCR results of different digestion time and ratios of templet and
primer:
{kind=link}
TRPC5 mutation region PCR
TPRC5 preserved bacteria plate streaking
DpnI digestion time experiment: “Whether 100ng plasmid can be digested completely by 1μl DpnI in 25μl, for 1h”
Result:
{kind=link}
25 th
Megaprimer PCR
- To find out the best annealing temperature for megaprimer. Set a series of annealing temperatures: 55, 60,65,70℃
- Repeat PCR
- Cycle pure to get purified PCR products.
- DpnI digestion
- Transformation
26 th
Megaprimer PCR results of different digestion time;PCR annealing temperatures and ratios of templet and primer:
{kind=link}
Colony number according to different annealing temperatures and ratios of templet and primer:
| PCR Annealing TEMP | Primer:Template= 1:1 | Primer:Template= 1:2 |
|---|---|---|
| 55℃ | 125 | 44 |
| 60℃ | 120 | 100 |
| 65℃ | 116 | 58 |
| 70℃ | 168 | 76 |
Sequencing results of T5 exonuclease digestion level according to time:
| T5 Exonuclease digestion time | 5' end digest number/bp | Direction |
|---|---|---|
| 5mins | 48 | Forward |
| 5mins | 43 | Reverse |
| 10mins | 176 | Forward |
| 10mins | 166 | Reverse |
Repeat T5 exonuclease digestion level according to time:
Set series of time gradients: 0, 5, 7.5, 10mins
a. T5 digestion (according to T5 exonuclease protocol)
| T5 Exonuclease | 3.4ul |
| NEB4.0 Buffer | 5ul |
| TRPC5 region | 3.4ug |
| ddH2O | to 50ul |
| Total | 50ul |
b. Take each group for Gel electrophoresis
{kind=link}
c. Send each group to sequence
Primer F:ATCCGAATTCCCCTCCAAATC
Primer R:TATGTTAAGTTCCCAGCCCAG
28 th
Sequencing results of T5 exonuclease digestion level according to time:
| T5 Exonuclease digestion time | 5' end digest number/bp | Direction |
|---|---|---|
| 5mins | 42 | Forward |
| 5mins | 43 | Reverse |
| 7.5mins | 104 | Forward |
| 7.5mins | 96 | Reverse |
| 10mins | 188 | Forward |
| 10mins | 156 | Forward |
29 th
Culture medium Pollution proving:
Set 4 groups of experiments (use sfGFP plasmid):
{kind=link}
30 th
Culture medium Pollution proving results:
Group 3 number > Group 1 number ; with lots of non-green bacteria
Group 4 has >100 colonies of non-green bacteria
Group 2 has no colony
Conclusion: Culture medium has been polluted.
Gibson assembly (pre-experiment)
1> Directly cut the mutation region by AflII and EcoRI for 8hrs
2> Gel extraction to get the 640bp fragment
{kind=link}
3> Use Gibson Assembly® Master Mix to get whole plasmid
4> Transformation
31 th
1.Gibson assembly result: NONE colony
2.Random mutagenesis with Kit, according to Random mutagenesis protocol
Sept.
01 st
Design qPCR Primers and Send for Synthesization
Sequences:
1.097&NeoLoxP qPCR-1st
{kind=link}
2. qPCR Internal Positive Control 553x118px
{kind=link}
04 th
Gibson Assembly
Insert : vector=2 : 3(50~100ng vector)
{kind=link}
T5 Exonuclease Digestion
Temperature: 50℃
{kind=link}
Transformation into Competent Cells
5μl product are transformed with 50ul competent cells.
Error-prone PCR(low frequency)
1. Information supplied with the GeneMorph II Random Mutagenesis Kit
(Agilent Technologies,Catalog #200550):
{kind=link}
2. Primers sequence:
Forward: ttactcaccatacagagatcgaattcc
Reverse: ttttccactttgctcagctccttaag
Target DNA mass: 500ng,corresponding to low frequency mutation
{kind=link}
4. Procedure
{kind=link}
The annealing temperature is set to be 57℃.
Gel Extraction of ep-PCR Product
Concentration: 135ng/μl, 30μl
qPCR Primer Test
Instrument: ABI StepOne Plus qPCR
Kit: Power SYBR® Green PCR Master Mix and RT-PCR Reagents Kit
Sample:
CHO cell genomic DNA extracted from 4.5x106 cells.
Concentration: 56.1ng/μl, 400μl
Target:
Neoloxp segment, 18sRNA, GAPDH
Standard:
097&Neoloxp plasmid(length:6635bp)
Concentration: 101.2ng/μl
{kind=link}
4.
{kind=link}
5. Procedure
{kind=link}
05 th
Transformation Result
No colony
Possible reason: The transformation efficiency of self-made competent cells is quite low.
06 th
TRPC5 PCR with PrimeStar
1.Primers sequence:
Forward: ttactcaccatacagagatcgaattcc
Reverse: ttttccactttgctcagctccttaag
2.Total 8 tubes of PCR reaction:
{kind=link}
3. Procedure
{kind=link}
Gel Extraction
{kind=link}
Concentration: 180ng/μl, 30μl, total 4 tubes.
T5 Digestion for 3.5 minutes and Cycle-pure
Concentration: 60ng/μl, 50μl
Mega-primer PCR with KOD Polymerase
Template DNA: 0.1ng,
Mega-primer:Template=10000:1
{kind=link}
Procedure:
{kind=link}
Cycle-pure and Gel Running
Concentration: 30ng/μl, 50μl
{kind=link}
No 6000bp band is appeared,it’s a problem!!
DpnI Digestion for 1 hour
Transformation
5μl product are transformed with 100ul competent cells.
qPCR Primer Test
Instrument: Bio-Rad
Kit: TaKaRa-One Step SYBR PrimeScript RT-PCR Kit I
1. Sample:
CHO cell genomic DNA extracted from 4.5x106 cells.
Concentration: 56.1ng/μl, 400μl
2. Target:
Neoloxp segment, 18sRNA, GAPDH
3. Standard:
097&Neoloxp plasmid(length:6635bp)
Concentration: 101.2ng/μl
{kind=link}
4.
{kind=link}
Wrong: Forget to add ‘PrimeScript Enzyme Mix’ which contains ‘Taq enzyme’,thus no PCR reaction works.
5. Procedure
07 th
Transformation Result
No colony
Possible reason: The mega-primer PCR procedure didn’t work.
Mega-primer PCR with Q5
1.Template DNA: 0.1ng,
Mega-primer:Template=10000:1
{kind=link}
2. Procedure
{kind=link}
qPCR Primer Test with Premix and Gel Running
3. Gel concentration: 2%(to better show the needed band)
{kind=link}
Procedure
{kind=link}
4. Results
{kind=link}
{kind=link}
The gel running results show that primer 1~4 and primer GAPDH has been contaminated,we need to use new primer and be care of experiment operation.
08 th
Gel Running with 30μl mega-primer PCR product
{kind=link}
There is a band at around 6000bp position!
DpnI Digestion for 1 hour
{kind=link}
Transformation
10μl product are transformed with 100ul competent cells.
9 th
Transformation Result
Little or no colony is appeared,maybe the transformation efficiency of competent cell is still too low.
09.12 Pre-experiment of enzyme digestion and ligation
TRPC5 PCR with PrimeStar
Primers sequence
Forward Primer: atccGAATTCccctccaaatc
Reverse Primer: ATGATCTCCAGTTCTCTGGA
2. Total 2 tubes PCR reaction
{kind=link}
3. Procedure
Gel Extraction
{kind=link}
Concentration: 44.4ng/μl, 40μl
EcoRI,SacI Restriction Enzyme Digestion
1. Insert
{kind=link}
The digestion is from 00:20 to 09:00.
2. Vector 300x214px
{kind=link}
The digestion is from 21:00 to 09:00.
qPCR Primer Re-design and Send for Synthesization
Sequences:
097&NeoLoxP qPCR-2nd
{kind=link}
13 th
Transformation
Use the bought competent cell,with transformation efficiency of approximately 10^7.
Gel Extraction of Enzyme Digested Vector
{kind=link}
Concentration of vector: 44.8ng/μl,20μl
Cycle-pure of Enzyme Digested Insert
Concentration of insert: 25.3ng/μl, 40μl
Ligation with T4 DNA Ligase
{kind=link}
The ligation is from 12:50 to 14:00.
Transformation
14 th
Transformation Result
{kind=link}
{kind=link}
{kind=link}
Competent Cell Efficiency
1. Formula: Colonies on plate / ng of DNA plated x 1000 ng/μg
2. Calculation: 237 / 0.02 x 1000=1.185x107 cfu/μg
3. Actual meaning: 20pg,5640bp→3.457x106 copies
4. 457x106 / 237=14586,which means 1 of 14586 vectors would actually grow on the plate.
Ligation Efficiency
1. 62ng,4906bp→1.232x1010 copies
2. Efficiency=
Mutation Library
1.Q5: 2.782x106
2.Gibson: 5.54x105
3. Ligation: 2.102x107
Repeat the pre-experiment on 09.12
Use enzyme digestion and ligation method to construct mutation library,except that using error-prone PCR,and the target DNA is 500ng.
{kind=link}
{kind=link}
15 th
qPCR Primer Test with Premix and Gel Running
1. Procedure is the same with the experiment on 09.07
2. Results:
{kind=link}
2. Conclusion:
I. Primer 1~4 are not good enough,cause they all can match with CHO cell’s gDNA and have PCR products.
II. Primer 2~4 are contaminated.
III. Primer GAPDH has unspecific PCR product,so it can’t be used.
Primer 18sRNA can be used.
18 th
Transformation
10μl positive ligation product are transformed with 125ul competent cells.For control group,5ul ligation product are transformed with 50ul competent cells.
qPCR Primer Test with Premix and Gel Running
1. We set 2 annealing temperature,which are 65℃ and 70℃ respectively.
2. Results:
865x276px3. Conclusion:
{kind=link}
I. Primer 1~4 are all not contaminated and have specific PCR band with annealing temperature of 65℃,while they don’t have PCR product band with annealing temperature of 70℃.
II. Raising annealing temperature can reduce unspecific amplification,while it can also decrease the PCR efficiency and cause Ct increasement.Also we need to verify whether the product band is what we need.
19 th
Transformation Result
{kind=link}
1. Mutation library(10ul ligation product)=10688
2. Ligation efficiency=
Pick 20 Single Colony for sequencing
Sequencing Result:
7 out of 20 plasmids have mutation(s) among specific region,in which one has 15 base mutations,one has 2 base mutations,and the rest five have 1 base mutation.
Scrape the Colonies for Endo-free Plasmid Extraction
The scraped colonies are cultured for around 9 hours till the OD achieves 2.879,with 150ml Amp.
Concentration: 55ng/μl, 1.75ml
20 th
qPCR Primer Test with Premix and Gel Running
1. We set 3 annealing temperature,which are 61℃,62℃ and 63℃ respectively.
2. Results:
21 th
CHO cell Genomic DNA Extraction
Positive: GECO+NFAT+Neoloxp
①d1-A10 ②d2-B5 ③d2-B9 ④d1-D3 ⑤d2-E9
⑥d1-F5 ⑦d1-F11 ⑧d1-A9 ⑨d1-B3 ⑩d2-C1
Control: GECO+NFAT
①C4 ②E6 ③F8 ④H10
22 th
qPCR Primer Test
Instrument: ABI StepOne Plus qPCR
Kit: TaKaRa-One Step SYBR PrimeScript RT-PCR Kit II
1. Sample:
CHO cell genomic DNA
Positive: transfected with GECO+NFAT+Neoloxp
Negative Control: transfected with GECO+NFAT
Wild Type Control: CHO cell genomic DNA
2. Target:
Neoloxp segment, 18sRNA
3. Standard:
097&Neoloxp plasmid(length:6635bp)
Concentration: 101.2ng/μl
{kind=link}
4.
5. Procedure
{kind=link}
6. Results 314x157px Conclusion
{kind=link}
I.Can’t use ‘TaKaRa-One Step SYBR PrimeScript RT-PCR Kit II’,cause our sample is DNA.
II.We forgot to add ‘PrimeScript Enzyme Mix’ at the beginning,which may cause inaccurate results.
III.We forgot to change pipette tips during the dilution,which may bring huge deviation.
IV.Two-step PCR has a low amplification efficiency in our experiment.To increase the efficiency,we need to use three-step PCR.
23 th
qPCR Primer Test
Instrument: ABI StepOne Plus qPCR
Kit: TaKaRa-SYBR Premix Ex Taq II(Tli RNaseH Plus)
1. Sample,Target and Standard are the same with the experiment yesterday.
2.
{kind=link}
3. Procedure 650x146px 4. Results 332x143px 471x117px 5. Conclusion
{kind=link}
{kind=link}
{kind=link}
Primer 3,4 are similarly efficient,combining with previous experiment result,we choose Primer 3 in the later experiment.
24 th
Repeat the mutation experiment on 09.14
1. The targeted mutation segments are 1ng,10ng,100ng respectively.
2. During the first PCR procedure,the annealing temperature is set to be 52℃,which is quite low,so it didn’t work.Then we change it to 57℃.
3. Procedure
{kind=link}
Gel Extraction of PCR Product(as Insert)
{kind=link}
Each with volume of 30μl.
EcoRI,SacI Restriction Enzyme Digestion for 9 hours
Cycle-pure
{kind=link}
qPCR Primer Test with Premix and Gel Running
Annealing temperature: 62℃
{kind=link}
Conclusion:
I.We can directly see the band’s brightness differences from the gel running result,which may indicate their plasmid copy number differences.Still,we need to do quantitative PCR to get more precise results.
II.It’s strange as well as unreasonable that band appears in wild-type control PCR result,which may indicate that it has been contaminated.
qPCR: 097-gDNA with Primer3
Instrument: ABI StepOne Plus qPCR
Kit: TaKaRa-SYBR Premix Ex Taq II(Tli RNaseH Plus)
Sample: Positive ①②③⑤⑦⑧⑩
Target: Neoloxp
Standard: 097&Neoloxp plasmid-③
2. Reagent and Procedure are the same with the experiment on 09.23.
3. Results
{kind=link}
{kind=link}
{kind=link}
4. Conclusion
Wild type control(new) may be contaminated.
25 th
We find that the synthesized TRPC5 has a problem,which may cause replacement during protein translation,so we need to re-construct TRPC5 plasmid.
Primer Synthesization(send to Invitrogen)
Sequences
Forward Primer: atccGAATTCccctccaaatc(we already have,don’t need to synthesize)
Reverse Primer: tgc tctaga gtg gcccagctgtactacaag
26 th
qPCR Primer Verification with Premix and Sequencing
Sequencing Results:
The PCR results are what we need,and the wild-type genomic DNA which were recently extracted are indeed contaminated.
27 th
Re-construct TRPC5 Plasmid
TRPC5 PCR with modify-primer
Same procedure as before
Gel Extraction
{kind=link}
XbaI,SacI Restriction Enzyme Digestion for Vector and Insert
28 th
Gel Extraction for Vector
Cycle-pure for Insert
T4 DNA Ligation for 20min
Transformation
After culture for 10 hours:
{kind=link}
Pick 8 Single Colony and Culture for Sequencing
Sequencing Result:
All correct.
Culture Colonies for Endo-free Plasmid Extraction
CHO Cell Genomic DNA Extraction
Positive: GECO+NFAT+Neoloxp
(11)d1-C1 (12)d1-C8 (13)d1-D12 (14)d1-E3 (15)d1-E4
(16)d1-F3 (17)d1-F6 (18)d1-F9 (19)d2-C3 (20)d2-C8
(21)d2-D5
29 th
PvuI,NcoI Restriction Enzyme Digestion for Verification
Verification result: All correct.
Oct.
01 st
Construct Mutation Library
Vector: TRPC5 digested with EcoRI,SacI,AflII
Insert: ep-PCR segments,with low&medium&high mutation frequency.
Gel Extraction of Enzyme-digested Vector
{kind=link}
Ligation for 1 hour
Transformation
5μl ligation product are transformed with 100ul competent cells.
qPCR: 097-gDNA with Primer3
Instrument: ABI StepOne Plus qPCR
Kit: TaKaRa-SYBR Premix Ex Taq II(Tli RNaseH Plus)
1. Sample: Positive (3),(11)~(21)
Negative: wild type cho cell gDNA
Target: Neoloxp
Standard: 097&Neoloxp plasmid( Endofree-① ),289ng/μl
The first and last dilution point are added with 10ng wild-type gDNA,to see its influence on standard curve.
2. Reagent and Procedure are the same with the experiment on 09.23,except that the annealing temperature is set to be 64℃.
3. Results
{kind=link}
{kind=link}
4. Conclusion
I. The plasmid concentration differs each time,we need to standardize the method for concentration measurement.
II. The primer is not good enough,which may cause inaccurate standard curve and copy number result.
02 nd
Scrape the Plate for Endo-free Plasmid Extraction
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Each with volume of 100μl.
CHO Cell Genomic DNA Extraction
Positive: GECO+NFAT+Neoloxp
d2-C6 (23)d2-A5 (24)d2-A8
d2-A9 (26)d1-D10 Wild type: CHO cell’s gDNA All the plasmids’ concentrations are re-measured using NanoDrop.
03 rd
qPCR: 097-gDNA with Primer3
Instrument: ABI StepOne Plus qPCR
Kit: TaKaRa-SYBR Premix Ex Taq II(Tli RNaseH Plus)
1. Sample: Positive (3),(22)~(26)
Negative: wild type cho cell gDNA(newly extracted)
Target: Neoloxp
Standard: 097&Neoloxp plasmid( Endofree-① , 307.8ng/μl
All the dilution points are added with 10ng wild-type gDNA.
2. Reagent and Procedure are the same with the experiment on 09.23,except that the annealing temperature is set to be 64℃.
3. Results
{kind=link}
{kind=link}
4. Conclusion
I. The standard curve efficiency is 140%,it may result from the adding of wild type gDNA in the preparation of standard point.
II. The Ct of wild type gDNA is 26,which indicates that it has been contaminated.So the copy number results are not precise,we can only compare their differences.