Difference between revisions of "Team:UESTC-software/Model"
| Line 22: | Line 22: | ||
<li><a href="https://2016.igem.org/Team:UESTC-software/Description">Description</a></li> | <li><a href="https://2016.igem.org/Team:UESTC-software/Description">Description</a></li> | ||
<li><a href="https://2016.igem.org/Team:UESTC-software/Design">Design</a></li> | <li><a href="https://2016.igem.org/Team:UESTC-software/Design">Design</a></li> | ||
| − | <li><a href="https://2016.igem.org/Team:UESTC-software/Features"> | + | <li><a href="https://2016.igem.org/Team:UESTC-software/Features">Features</a></li> |
<li><a href="https://2016.igem.org/Team:UESTC-software/Model">Model</a></li> | <li><a href="https://2016.igem.org/Team:UESTC-software/Model">Model</a></li> | ||
| Line 60: | Line 60: | ||
</header> | </header> | ||
<div class="content-top"> | <div class="content-top"> | ||
| − | <img src="https://static.igem.org/mediawiki/igem.org/ | + | <img src="https://static.igem.org/mediawiki/igem.org/9/9a/Uestc_software-project_modeling2.png"> |
<p class="title">Model</p> | <p class="title">Model</p> | ||
</div> | </div> | ||
Revision as of 07:45, 19 October 2016

Model
Our project is mainly centered on how to improve the storage density and avoid the mistakes that may occur during the process of designing a stable, high-density DNA information storage system. There are two main technological processes, encoding and decoding, in the system.
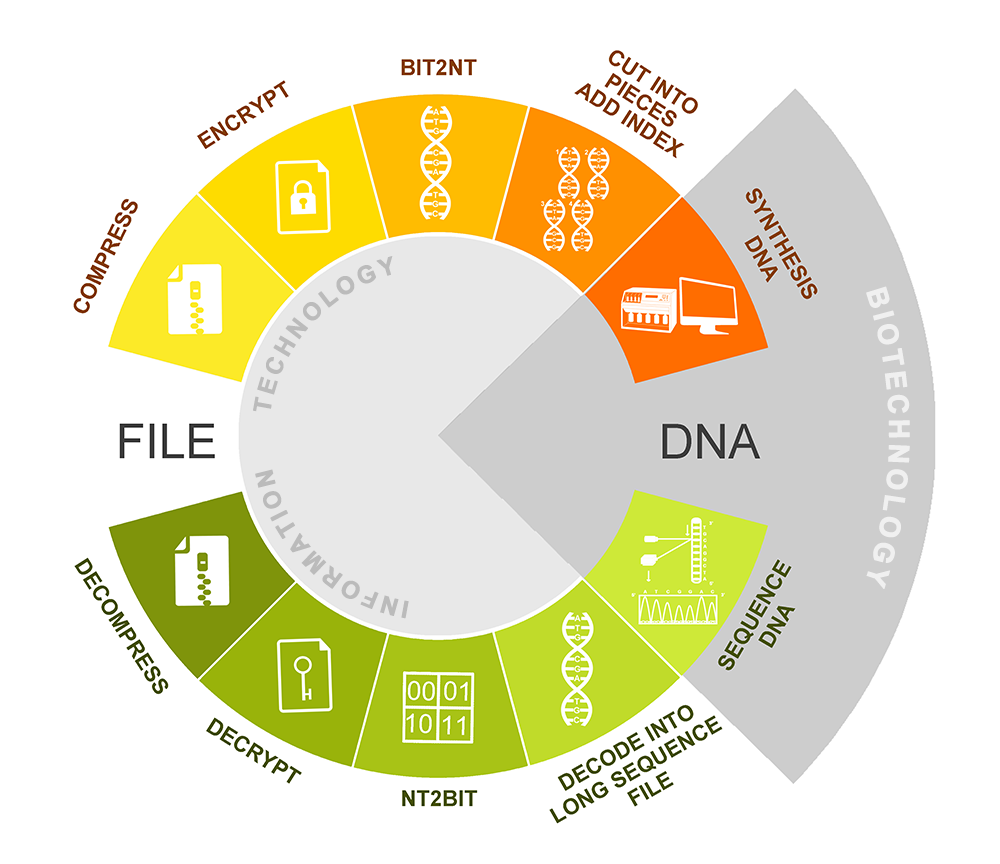
Fig.1.system flow diagram.
Encoding
Compression: ZIP algorithm
We used bzip2 algorithm which renowned as a high-quality data compression algorithm to compress the file. It typically compresses files to within 10% to 15% of the best available techniques, whilst being around twice faster at compression and six times faster at decompression. After this process, we get a bz2 compression file.
Encryption: ISAAC encryption algorithm
Next, we use ISAAC encryption algorithm to encrypt the bz2 file. After you input your own password, ISAAC generates a pseudorandom stream of bits (a keystream). As with any stream cipher, these can be used for encryption by combining it with the plaintext using bit-wise exclusive-or; decryption is performed the same way (since exclusive-or with given data is an involution). After this process, we can get a sufficiently random binary file.
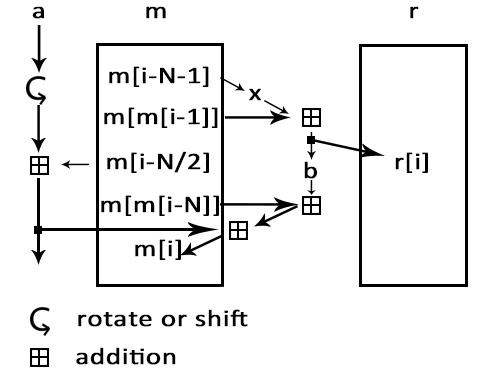
Fig.2. encryption process.
Bit-to-nt conversion: quanternary system
In bit-to-nt conversion process, we use the idea of the quaternary system. As we all know, there are four basic groups—A, T, C, G, and it can be seen as a quaternary system. In bit-to-nt conversion, one byte of bits converts into four bytes of A, T, C, G, using the scheme illustrated in Table 1.
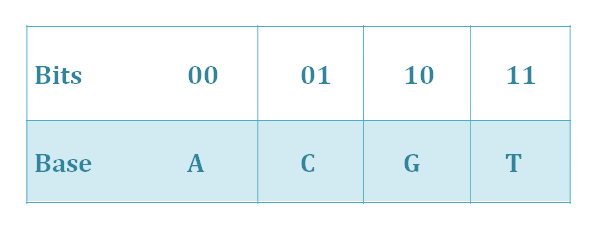
Tab.1.Bit-to-nt conversion.
We read out the binary string S0 of the binary file generated in the last process. Use bit-to-nt conversion to convert S0 into a DNA string S1. We get a long DNA sequence.
Fragmenting & indexing
Write len( ) for the function that computes the length of a string, and define n=len(S1). Represent n in base-4 and prepend ‘0’s to generate a string S2 of quaternary such that len(S2)=15. Form the string concatenation
S4=S1.S3.S2(1)
(the symbol ‘ . ’ means the connection of two srings)
where S3 is a string of at most 49 ‘ 0 ’s chosen so that len(S4) is an integer multiple of 50.
Convert S2 and S3 to DNA strings S2’ and S3’using the scheme illustrated in Table 2.

Tab.2.Quaternary-to-nt conversion.
Recode the DNA string S3’.S2’ from the second character to S2’’ with repeated nucleotides as few as possible using the scheme illustrated in Table 3.

Tab.3.Recoding table.
From
S5=S1.S2’’(2)
Even-odd check
Define N=len(S5). Split S5 into overlapping segments of length 200 nt, each offset from the previous by 50 nt. This means there will be (N/50)-3 segments, conveniently indexed
i = 0,1,......,(N/50)-4(3)
segment i is denoted Fi and contains (DNA) characters
50i,......,50i199(4)
of S5. If i is odd, reverse complement Fi.
Let i4 be the base-4 representation of i, appending enough leading ‘0’s so that
len(i4) = 12(5)
Recode i4 using the same strategy in Table 2 & Table 3 above, and i4 is represented in nt.
Compute P as the sum(mod 4) of the even-positioned quaternary in i4.
P = ([i4]2[i4]4[i4]6[i4]8[i4]10[i4]12 ) mod 4(6)
P acts as a ‘parity quaternary’—analogous to a parity bit—to check for errors in the encoded information about i.Form the indexing information string
IX = i4.P(7)
(comprising 121=13 nt)
Append the DNA-encoded version of IX to Fi to give indexed segment Fi’.
Then form Fi’’ by prepending A or T and appending C or G to Fi’ —choosing between A and T, and between C and G, randomly if possible but always such that there is no repeated nt. This ensures that we can distinguish a DNA segment that has been reverse complemented during DNA sequencing from one that has not—the former will start with G|C and end with T|A; the latter will start A|T and end C|G.
The segment Fi’’ are synthesized as actual DNA oligonucleotides and stored, and may be supplied for sequencing.
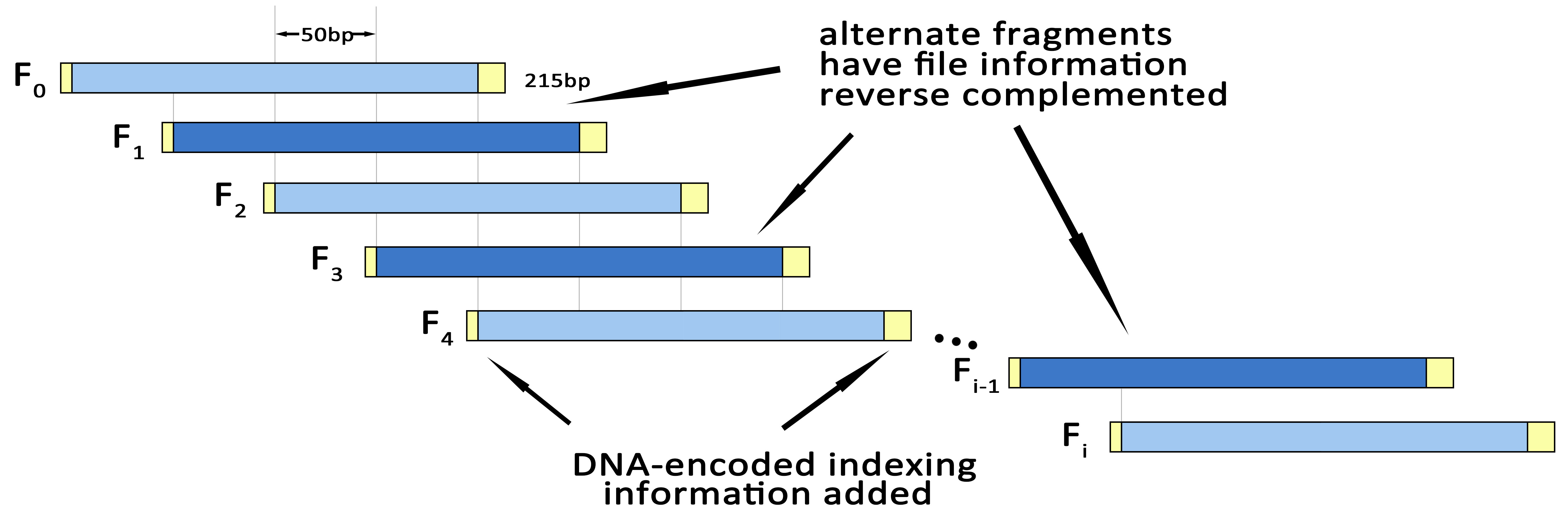
Fig.3.Fragmenting & Indexing.
Safety testing: BLAST
In order to check whether the sequences we generated are safe, we use BLAST to compare the sequences with the Biobricks database. We use all the Biobricks sequences to establish a FASTA format file, and use BLAST to format it to set up a BLAST database. Then through local BLAST, we compare the sequences generated with the Biobricks sequences to confirm that the sequences are out of bio-function.
Decoding
Recognition of the front and the end of a sequence
Reverse complementation during the DNA sequencing procedure (e.g. during PCR reactions) can be identified for subsequent reversal by observing whether fragments start with A|T and end with C|G, or start with G|C and end with T|A.
Reading and check of index
With these two ‘ orientation ’ nt removed, the remaining 213 nt of each segment can be split into the first 200 ‘ message ’ nt and the remaining 13 ‘indexing’ nt. Decode the ‘ indexing ’ nt to quanternary using the reverse of the encoding tables in in Table 2 & Table 3 above, and use P to check the correctness of i4.
Correction of segments through four-fold redundancy
Use i to determine the location of each fragment. Split each fragment into segments of length 50 nt. As we provide a four-fold redundancy, we compare the 50-nt segments which are in the same location and inaccurate bases can be corrected by using majority vote. Connect all the segments to a whole DNA sequence.
Decoding, decryption and decompression
Decode the DNA sequence by using the reverse of the encoding table in Table 1 above, and use ISAAC to decrypt it, and then decompress. We get the original file.
Fuzzy matching with high-throughput sequencing
Errors introduced during DNA synthesis, storage or sequencing could lead to various nt insertion, deletion or substitution. Recovery of information from fragments with such errors may be possible via PCR amplification and high-throughput sequencing.
Example
In order to make our encoding process more understandable and clearer, we show an example here.
With a given text file, we read out its binary file, then compress it with bzip2 algorithm, and use a pseudorandom stream of bits to encrypt it. Then use bit-to-nt conversion to convert the binary numbers to bases. Shown in Figure 4.

Fig.4.Schematic of DNA information storage system.
The computer file (in any format, e.g. text) shown in (a) is read in binary format (b), and use bzip2 to compress it (c). Then through ISAAC encryption algorithm use pseudorandom stream of bits (d) and exclusive-or to generate a new binary string (e). Then one byte of bits converts into four bytes of A, T, C, G (e).
1. Towards the text file “IGEM UESTC-SOFTWARE”, its binary coding is

2. After compressing, its compressed coding is:

3. With the pseudorandom stream of bits that ISAAC64 generated (password: uestc-software):
(lack of data, data need to be caculate)
Using XOR operation:
S’ XOR (pseudorandom stream of bits)
We get a new binary string S0:

4. Using the scheme illustrated in Table 1, we convert the bytes of S0 into bases—A, T, C, G.

5. n=len(S1 )=3376, which is 310300 in base-4. So:
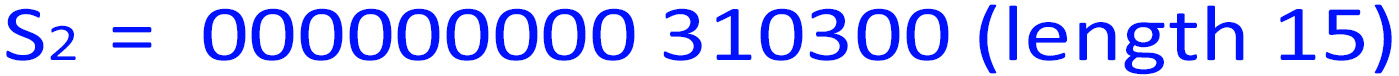
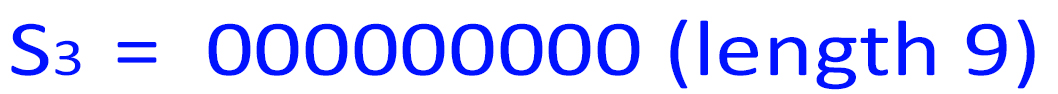
len(S4 )=len(S1 )len(S2 )len(S3 ) = 3376 + 15 + 9 = 3400 = 50 * 68
6. Using the scheme illustrated in Table 2, convert S2 and S3 to DNA:

Recode the DNA string S3’ . S2’ from the second character to S2’’ with repeated nucleotides as few as possible, using the scheme illustrated in Table 3.

7. N=len(S5 )=3400. We split S5 into overlapping segments of length 200 nt, each offset from the previous by 50 nt.
S5 will be split into overlapping segments Fi of length 200 nt for
i=0......(3400/50)-4, i=0,1,......,64
With overlapping parts underlined for illustration, F0 to Fi are:

8. Only i=1,3,...,63 are odd, so Fi is reverse complemented:

9. For i=0,i4=000000000000 (length 12) and the sum (mod 4) of the even-positioned quaternaries of i4 is
P = (0+0+0+0+0+0)(mod 4) = 0
For i=1,i4=000000000001
P = (0+0+0+0+0+1)(mod 4) = 1
For i=0,IX=i4.P=0000000000000
For i=0,IX=i4.P=0000000000011.
So:

11. Prepend A|T and append C|G (in this example we have three random choice, at the front of F0’’, the end of F0’’, and the end of F1’’):


 CATALOGUE
CATALOGUE