Team:Tianjin/Experiment/Protein Engineering
Protein Engineering
Rational Design
Motivation
The degradation of PET is completed in two steps by PETase and MHETase. PETase hydrolyze PET to mono(2-hydroxyethyl) terephthalic acid (MHET), which will be further decomposed by MHETase into two monomers, terephthalic acid (TPA) and ethylene glycol (EG)[1] . In this two-step reaction system, MHET, the product of PETase-mediated hydrolysis of PET, was found to be a very minor component, which reveals rapid MHET metabolism[1], indicating the rate determining step in this reaction is the first step, hydrolysis of PET. So in order to accelerate the whole PET degradation speed, increasing the activity of PETase is rather crucial. To enhance PETase hydrolysis activity, we first tried to understand the mechanism of the hydrolysis reaction by generally confirming active sites of PETase and 3 dementional structure simulation.
Serine-based Catalytic Triad Mechanism & 3D Model Simulation
Since there is no x-ray structure for PETase , the mechanism of PETase hydrolysis activity can not be exactly identified. But the binding site and catalytic site can be generally inferred according. Based on the efforts have been made to identify and characterize bacterial cutinases[2], α/β hydrolase fold family contain a highly conserved characteristic GXSXG motif. With sequence analysis, PETase was also found to contain an accordant GWSMG motif.
So we simulated a best fit model for PETase by SWISSMODEL, an automated comparative protein modeling server[3]. The template was Thc_Cut2, which shares 52% sequence identity with PETase[4]. As expected, the homology model of PETase displays a canonical α/β hydrolase fold with a Ser160-His237-Asp206 catalytic triad and a preformed oxyanion hole (Fig.1), suggesting a classic serine hydrolase mechanism.

Ribbon diagram of a predicted PETase model. The catalytic triad residues are shown as ball-and-sticks in green, formed by Ser160, His237 and Asp206, and the oxyanion hole binding site residues are in blue, formed by the main chain amides of Met161 and Tyr87.
Mutation Design Rationales
1.Hydrophobicity & space factor
Given that the hydrolysis of PET is a heterogeneous reaction, a prerequisite for its efficient reaction is an adequate interaction of the enzyme with the substrate at the solid-liquid interface. Since the substrate PET is hydrophobic polymer, both the hydrophobicity of the enzyme surface and the space around active site to accommodate PET may be the main factors for more efficient protein attachment and reaction activity[6]. In fact, tailoring enzymes for a more hydrophobic and larger substrate active site has been shown to facilitate PET hydrolysis by a fungal polyester hydrolase[5]. Also Silva et al. obtained a double mutant (Q132A/T101A) exhibiting a 2-fold increased hydrolysis activity against PET fibers by the substitution of spacious residues with alanine in its substrate binding site[6]. So we aimed to take the same strategy to modify hydrophobicity of the surface of PETase as well as broaden space near the active site to enhance the interaction between PETase and substrate.
2.Electrostatics factor
The electrostatic surface property in the vicinity to the active site was also found to be responsible for differences in hydrolysis efficiency. Exchange of the positively charged amino acids to the non-charged ones strongly increase the hydrolysis activity. In contrast, exchange of the uncharged amino acids by the negatively charged ones can lead to a complete loss of hydrolysis activity on PET films[7]. Changing the surface property is another strategy we took.
3.Conserved Residues
In another study, Wei and coworkers exchanged selected amino acid residues of TfCut2 involved in substrate binding with those in LC-cutinase(LCC) to relieve product inhibition and obtained enzyme variants with increased PET activity at 65℃ [8]. In the inspiration of their work as well as that the position of catalytic triad hold constant in different mature PET hydrolases with signal peptide excluded, we exchanged amino acid residues on the surface with highly conserved residues of the same position in other PET hydrolases, which can be a way to either increase the enzyme efficiency or confirm the essential sites which made PETase more efficient than other PET hydrolase.
Mutants
Based on the above rationales, our general strategies of mutation design are as follows:
1).According to the 3D structure of PETase we simulated, which shows the hydrophobicity of protein surface (Fig.2), we changed the hydrophilic amino acid residues on the surface with hydrophobic ones to enhance the surface hydrophobicity;
2). Modify the electrostatics of its surface by changing charged amino acid residues to the uncharged ones;
3). Based on 3D structure predicted and docking simulation with substrate, we found several spacious residues adjacent to active site and binding site, which may inhibit the interaction of protein and substrate due to its large body. We substituted them with smaller amino acids to expose the catalytic site and binding site;

The deeper the bule is, the more hydrophobic the protein surface is; On contrast, the deeper the brown is, the more hydrophilic the protein surface is.
4). We generated a multiple sequence alignment by ClustalX with seven serine hydrolases from NCBI conserved with GXSXG motif and catalytic triad and reported to be able to degrade PET (Table.1). Based on the multiple sequence alignment result(Fig.3), we exchanged some essential residues adjacent to active site and binding site with the conserved residues of the same position in the multiple sequences.

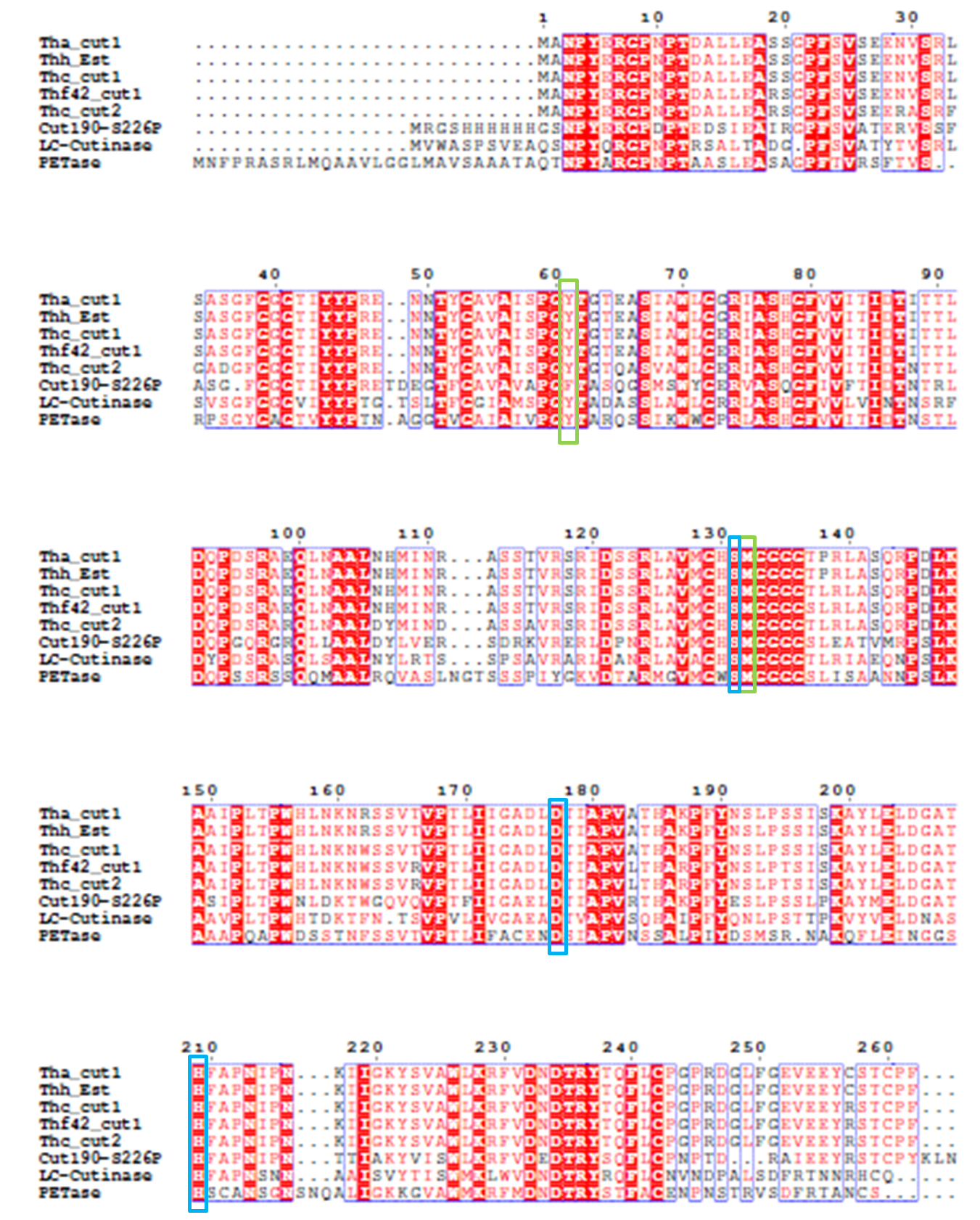
The catalytic triad is shaded in blue box and the binding site is shaded in green box.
The position of catalytic triad hold constant throughout the multiple sequences
in mature protein with signal peptide excluded.
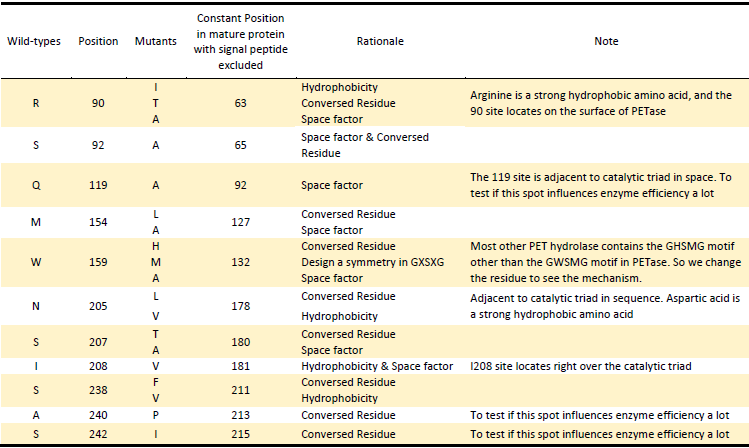
Based on the Rationales above, we designed 22 potential mutations
in purposes to enhance the efficiency of PETase hydrolysis activity
and confirm potential determining site of PETase hydrolysis.
Site-directed mutagenesis
Site-directed mutagenesis was performed using recombinant PCR technique[9]. Our modified approach is based on the PCR amplification of target PETase fragment by mutagenic primers divergently oriented but overlapping at their 5’ ends. The mutagenic nucleotides are located in both forward and reverse primers. The approach contained two rounds of PCR. In the PCR Round I, we use PETase fragment as template, respectively use original forward primer of PETase & mutagenic reverse primer, and original reverse primer & mutagenic forward primer as primers. The product of PCR Round I are portions of PETase gene with an overlapping region. In the PCR Round II, we use products of PCR Round I as two templates and original primers as primers to generate the final mutant fragment (Fig.4). And each specific mutation was verified by sequencing.

High-troughput Assay Strategy——CFPS
Motivation
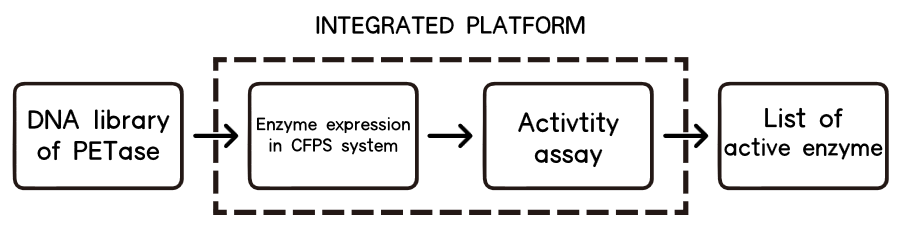
Protein Engineering aiming at increasing protein activity by rational design has been an common occurrence for years. With the knowledge of protein structure as well as catalytic mechanism[11], specific changes are made in an attempt to enhance the function of the protein. Quite a few literatures have reported improvement enzyme activities of different functionby rational design[5-10]. Also, some prior iGEM teams tried to imply various different approaches to design and generate mutations as well.
However, one of the main bottlenecks for it is an expression and assay method, which can be easy-to-implement and especially fast enough for high-throughput screening. In our case, we need to find a fast way to do the assay and an ideal chassis to express or even secret our twenty–two mutations.

E.coli has been one of the most widely used modified chassis, however, the traditional way of cell breakage for protein extraction[12] along with further purification is time-consuming and low-efficient. Saccharomyces cerevisiae is good for secretion, and was the first chassis we tried to transform our mutations. But due to its relatively slow growing speed and laborious transformation process, it’s not suitable for a large scale of mutation selection.
After intensive research, we finally decided that no chassis is needed, the best way for us to implement high-throughput assay to select mutations is a cell-free system for its rapid one-pot expression[13], capability of being analyzed without extensive purification[14], and real-time monitoring of protein expression as a fluorescence-based approach.
Cell-free Protein Synthesis(CFPS)
Cell-free protein synthesis (CFPS), is the production of protein from nucleic acid templates in the test tube without the use of living cells. Thus CFPS enables direct access and control of the transcription and translation environment which is benefit for specific reactions.
CFPS is programmed by addition of a DNA template, formed from either closed circular vector DNA or a linear PCR product[15]. Common components of a CFPS system include cell extract, the needed energy source, along with a feeding solution[16] (which includes substrates such as amino acids, ATP and GTP), and cofactors such as magnesium. A cell extract is obtained by lysing the cell of interest and centrifuging out the cell walls, DNA genome, and other debris. The remains are the cell machinery including ribosomes, RNA-Polymerase and etc., which are necessarily needed to function properly. Transcription is performed by recombinant phage T7 RNA polymerase (RNAP), generating the mRNA upon which the ribosomal translation machinery acts[17].
CFPS has many advantages over the traditional in-vivo synthesis of proteins. The open nature of CFPS allows direct manipulation of the chemical environment so as to optimize functional protein synthesis and concentrations, as well as control the reaction process. In contrast, once DNA is inserted into live cells, the reaction cannot be accessed until it is over and the cells are lysed.
High-throughput Selection
CFPS, especially is an ideal strategy for high-throughput selection and assay. For the first time, researchers have been able to express and purify a large number of proteins in a short period of time for subsequent high throughput functional and structural analyses[14]. And the use of improved fluorescent proteins and of fluorescence detection technologies in a plate reader platform, allow real time monitoring of protein expression in a high-throughput format.
Most notably, CFPS system is much faster than in-vivo synthesis. To express our mutation, a cell free reaction, including extract preparation, usually takes only a few hours, whereas in-vivo protein expression (in yeast) takes 3-5 days including transformation and incubation, and it’s even longer if the promoter is an inductive one.
Not to mention protein purification, researchers had to individually purify proteins and spot each protein on a solid surface using conventional methods[18]. These protein arrays are laborious to make, and surface-bound proteins can lose functions during storage. Cell-free protein synthesis circumvents these problems and can be analyzed without extensive purification.

Experiment Design
Basically, we utilized the cell-free system to express the enzymes which had been modified in 22 different sites. Besides, we added a fluorescet protein, CFP, before the enzyme. And there is a flexible linker, GGGGSGGGGS , between them. So we could detect the expression of enzymes by detecting expression of the fluorescent protein with a fluorescence readout instrument, for example, a microplate reader. We conceived that with this method we could acquire the best modifications by screening them in a high-throughput way. Then we used the proteins we got to degrade PET.
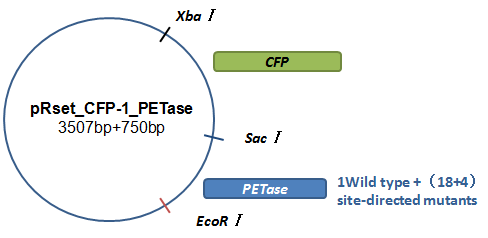
How to characterize the degradation velocity is the main problem in our scheme. We analyzed the experiment consequences in two ways. For the first one, we rendered the enzymes degrade pNPa, a general substituent for the detection of PET. Then we measured the absorbance of pNP in the optical density of 400 nanometers, which is the degrading product of pNPa. For the second one, we detected the absorbance of MHET in the optical density of 260 nanometers, which is the product in the first step of PET degradation.
Click here to find the
CFPS Protocol
We attempted to use two substrate for our mutant assay, one is PET film and the other is p-nitrophenyl acetate (pNPA). pNP-aliphatic esters is a kind of universal assay substrate of PET degradation activity for its rapid reaction and visible color of hydrolysis product. However, as pNPA is soluble, the using of pNPA may not allow us to select out the more efficient mutant improved by being designed to enhance the surface hydrophobicity. Most notably, according to Yoshida et.al, the assay result of activity comparison between PETase, LC cutinase(LCC), and F.solani cutinase(FsC) shows that the activity for pNP-aliphatic esters of PETase is lower than that of LCC and FsC, however, the activity of PETase against PET film is 5.5 and 88 times as high as that of LCC and FsC[1]. Hence aiming at breaking PET plastic and solving real-condition problem, we finally decided to use PET film as our substrate and detect the hydrolysis product mono(2-hydroxyethyl) terephthalic acid (MHET) at an absorption of 260nm. Due to lacking of MHET standard (which is not a commercial agent), we failed to draw calibration curve of MHET at 260nm. So we compared the activity of mutants and wild-type with unit absorption, which is positively related to concentration of the detecting product.
References
[1] Yoshida S, Hiraga K, Takehana T, et al. A bacterium that degrades and assimilates poly (ethylene terephthalate)[J]. Science, 2016, 351(6278): 1196-1199.
[2] Chen S, Tong X, Woodard R W, et al. Identification and characterization of bacterial cutinase[J]. Journal of Biological Chemistry, 2008, 283(38): 25854-25862.
[3] Schwede T, Kopp J, Guex N, Peitsch MC. SWISS-MODEL: an automated protein homologymodeling server. Nucleic Acids Research, 2003, 31 (13): 3381-3385
[4] Yoshida S, Hiraga K, Takehana T, et al. Supplementary Material For A bacterium that degrades and assimilates poly (ethylene terephthalate)
[5] Araújo R, Silva C, O’Neill A, et al. Tailoring cutinase activity towards polyethylene terephthalate and polyamide 6, 6 fibers[J]. Journal of Biotechnology, 2007, 128(4): 849-857.
[6] Silva C, Da S, Silva N, et al. Engineered Thermobifida fusca cutinase with increased activity on polyester substrates[J]. Biotechnology journal, 2011, 6(10): 1230-1239.
[7] Herrero Acero E, Ribitsch D, Dellacher A, et al. Surface engineering of a cutinase from Thermobifida cellulosilytica for improved polyester hydrolysis[J]. Biotechnology and bioengineering, 2013, 110(10): 2581-2590.
[8] Wei R, Oeser T, Schmidt J, et al. Engineered bacterial polyester hydrolases efficiently degrade polyethylene terephthalate due to relieved product inhibition[J]. Biotechnology and bioengineering, 2016.
[9] Ansaldi M, Lepelletier M, Méjean V. Site-specific mutagenesis by using an accurate recombinant polymerase chain reaction method [J]. Analytical biochemistry, 1996, 234(1): 110-111.
[10] Roth C, Wei R, Oeser T, et al. Structural and functional studies on a thermostable polyethylene terephthalate degrading hydrolase from Thermobifida fusca[J]. Applied microbiology and biotechnology, 2014, 98(18): 7815-7823.
[11] Wilson C J. Rational protein design: developing next‐generation biological therapeutics and nanobiotechnological tools[J]. Wiley Interdisciplinary Reviews: Nanomedicine and Nanobiotechnology, 2015, 7(3): 330-341.
[12] Kamioka T, Sohya S, Wu N, et al. Extraction of recombinant protein from Escherichia coli by using a novel cell autolysis activity of VanX[J]. Analytical biochemistry, 2013, 439(2): 212-217.
[13] Carlson E D, Gan R, Hodgman C E, et al. Cell-free protein synthesis: applications come of age[J]. Biotechnology advances, 2012, 30(5): 1185-1194.
[14] Chong S. Overview of Cell‐Free Protein Synthesis: Historic Landmarks, Commercial Systems, and Expanding Applications[J]. Current Protocols in Molecular Biology, 2014: 16.30. 1-16.30. 11.
[15] Whittaker J W. Cell-free protein synthesis: the state of the art[J]. Biotechnology letters, 2013, 35(2): 143-152.
[16] Rosenblum G, Cooperman B S. Engine out of the chassis: cell-free protein synthesis and its uses[J]. FEBS letters, 2014, 588(2): 261-268.
[17] Beckert B, Masquida B. Synthesis of RNA by in vitro transcription[J]. RNA: Methods and Protocols, 2011: 29-41.

