|
|
| (18 intermediate revisions by 2 users not shown) |
| Line 3: |
Line 3: |
| | {{:Team:Tianjin/Templates/AddCSS|:Team:Tianjin/Experiment/R-R/style.css}} | | {{:Team:Tianjin/Templates/AddCSS|:Team:Tianjin/Experiment/R-R/style.css}} |
| | {{:Team:Tianjin/Templates/AddCSS|:Team:Tianjin/Community/css/bootstrap.css}} | | {{:Team:Tianjin/Templates/AddCSS|:Team:Tianjin/Community/css/bootstrap.css}} |
| − | {{:Team:Tianjin/Templates/AddCSS|:Team:Tianjin/Community/css/font.css}}
| |
| − | {{:Team:Tianjin/Templates/AddCSS|:Team:Tianjin/Community/css/animation.css}}
| |
| − | {{:Team:Tianjin/Templates/AddCSS|:Team:Tianjin/Community/css/animate.css}}
| |
| | | | |
| | <html> | | <html> |
| Line 46: |
Line 43: |
| | <h1 id="about" class="title text-center">Experiment of <span>CFPS</span></h1> | | <h1 id="about" class="title text-center">Experiment of <span>CFPS</span></h1> |
| | <h2><b>Overview</b></h2> | | <h2><b>Overview</b></h2> |
| − | <p style="font-size:18px">R-R system (namely reporting and regulation system), is used in our project in order to make the expression of PET degrading enzyme visible and regular. As its name implies, this system consists of two independent part, reporting and regulation. We test our reporting part in <i>E.coli</i> and regulation part in <i>Saccharomyces cerevisiae</i> .</p> | + | <p style="font-size:18px">The CFPS system is the assay system in our project for the enzyme modification. The ability to produce a functional protein in the test tube, rather than in cells, is the essence of cell-free protein synthesis (CFPS)[1].</i> .</p> |
| | + | <div class="row"> |
| | + | <div class="col-md-2"></div> |
| | + | |
| | + | |
| | + | <div class="col-md-8"> |
| | + | <img src="https://static.igem.org/mediawiki/2016/5/5e/T--Tianjin--cf-e-1.png" alt="desktop"> |
| | + | <p style="font-size:15px"> |
| | + | <br/> |
| | + | Fig.1. The basic process of CFPS system |
| | + | </p> |
| | + | <div class="col-md-2"></div> |
| | | | |
| − |
| |
| − | <div class="row">
| |
| − | <div class="col-md-3">
| |
| − | <br/><br/><br/><br/>
| |
| − | <img src="https://static.igem.org/mediawiki/2016/3/32/T--Tianjin--R-R_system1.jpg" alt="desktop">
| |
| − | <br/><p style="font-size:15px">Fig.1. Structure of part <i><i>BBa_K339007</i></i></p>
| |
| − | <div class="space"></div>
| |
| − | </div>
| |
| − | <div class="col-md-9">
| |
| − | <h3><b>1. Reporting System</b></h3>
| |
| − | <p style="font-size:18px">The basis of our reporting system is the part <i><i>BBa_K339007</i></i>, Designed by Emily Hicks from Group iGEM10_Calgary. This part can sense the CpxR protein, which will form spontaneously in <i>E.coli</i> when inclusion body and misfolding protein present in the periplasm of <i>E.coli</i>, and then start expressing RFP so that we can detect red fluorescence. As we all know, the inclusion body will inevitably form when we overexpress heterologous protein like PETase in <i>E.coli</i>. Therefore, the emission of red fluorescence can report the overexpression of PETase. What is more, this device can be modified to report overexpression of any heterologous protein only if the PETase gene is replaced by another heterologous gene. After the red fluorescence is detected, we could start the purification of protein.</p>
| |
| | | | |
| | </div> | | </div> |
| − | </div>
| + | |
| − |
| + | </div> |
| − | <div class="row"> | + | <p style="font-size:18px">The lack of high-throughput approaches for expression and screening of large enzyme libraries is a |
| | + | major bottleneck for current enzyme engineering efforts. To address this need, some researchers[2] have developed a high-throughput, fluorescence-based approach for rapid one-pot, microscale expression, and screening of |
| | + | different kinds of enzymes. To go further, we try to make our effert to achieve integration of cell-free protein expression with activity screening of enzymes( site-directed mutants of <i>PETase</i>). |
| | + | </i> .</p> |
| | + | <div class="row"> |
| | <div class="col-md-2"></div> | | <div class="col-md-2"></div> |
| | | | |
| | | | |
| | <div class="col-md-8"> | | <div class="col-md-8"> |
| − | | + | <img src="https://static.igem.org/mediawiki/2016/9/9c/T--Tianjin--cf-e-2.png" alt="desktop"> |
| − | <img src="https://static.igem.org/mediawiki/2016/9/96/T--Tianjin--R-R_system2.jpg" alt="desktop"> | + | |
| | <p style="font-size:15px"> | | <p style="font-size:15px"> |
| | <br/> | | <br/> |
| − | Fig.2. Brief Structure of our reporting system based on inclusion body sensing CpxR promoter | + | Fig.2.One-pot approach for integrated expression and activity screening of enzymes |
| | </p> | | </p> |
| − |
| |
| | <div class="col-md-2"></div> | | <div class="col-md-2"></div> |
| | | | |
| Line 81: |
Line 80: |
| | | | |
| | </div> | | </div> |
| − |
| |
| | | | |
| − | <div class="row"> | + | <h2><b>Background</b></h2> |
| − | <div class="col-md-5">
| + | <h3>a.Synthetic Biology</h3> |
| − | <h3><b>2. Cell Lysis Based Regulation System</b></h3>
| + | <p style="font-size:18px">At its core, synthetic biology is inspired by the power and diversity of the living world. What is unique to synthetic biology is the application of an engineering-driven approach to accelerate the design-build-test loops required for reprogramming existing, and constructing new, biological systems. However, a major challenge exists because of our incomplete knowledge of how life works, the daunting complexity of cells, the unintended interference between native and synthetic parts, and (unlike typical engineered systems) the fact that cells evolve, have noise, and have their own agenda such as growth and adaptation[3].In addressing this questions, cell-free systems will play a very inportant role. |
| − | <br/> <p style="font-size:18px">The regulation system consists of two section. The first section is based on the already mentioned reporting system. We change the RFP gene to the novel ddpX (D-alanyl-D-alanine dipeptidase) gene from <i>E.coli</i> genome. The ddpX gene can hydrolyze the D-Ala-D-Ala structure in peptidoglycan molecule and cause damage to the cell wall of <i>E.coli</i>. Under normal condition, this gene only express when the cell is in starvation mode in order to use hydrolysate alanine as carbon source. However, if we overexpress this gene, the cell wall will be dissolved and finally cell lysis will happen. Therefore, in this system, when the PETase is overexpressed, the spontaneously forming inclusion body will induce the expression of ddpX and cause cell lysis. It will provide us with a novel and convenient and way of protein purification when you use <i>E.coli</i> as chassis.</p>
| + | |
| − | </div>
| + | |
| − | <div class="col-md-7">
| + | |
| − | <br/><br/><br/><br/><br/><br/><br/><br/>
| + | |
| − | <img src="https://static.igem.org/mediawiki/2016/thumb/9/96/T--Tianjin--R-R_system3.jpg/800px-T--Tianjin--R-R_system3.jpg" alt="desktop">
| + | |
| − | <p style="font-size:15px"> Fig.3. Brief Structure of our regulation system based on cell lysis</p>
| + | |
| − |
| + | |
| | | | |
| − |
| + | </p><br/> |
| − | </div>
| + | <h3>b.The system</h3> |
| − | </div>
| + | <p style="font-size:18px">Today cell-free protein synthesis(CFPS) has become a widely used method in molecular biology. In addition to the challenges in vivo above, production of proteins using cell-free protein synthesis usually takes only a few hours, in contrast to production of proteins in cells, which typically takes days to weeks. In fact, even first-time users can often obtain newly synthesized proteins in one day using a commercial system.</p><br/> |
| − |
| + | |
| − | <div class="row"> | + | |
| − | <div class="col-md-5">
| + | |
| | | | |
| | + | <h3>c.Theoretic background</h3> |
| | + | <p style="font-size:18px">click here to find the<a href="https://2016.igem.org/Team:Tianjin/Protocol">CFPS Protocol</a> |
| | + | The compounds we will talk about are in the Feeding Buffer of the Protocol, which also plays the role of buffer solution. |
| | + | For the transcription peorid, from tamplate DNA to mRNA, the necessties including 4 monosomes of RNA, RNA polymersase, energy resourse and other ions. Besides, our CFPS system including the lysate derived from Escherichia coli extract. |
| | + | The translation machinery is the engine of life. In order to synthesis the protein successfully, the 20 amino acids and ribosomes are obviously necessary. In E.coli CFPS the translation machinery is typically about 20-fold more dilute than in cell, decreasing the rates of initiation, elongation and protein accumulation . As well, the average distance between two adjacent ribosomes on a single mRNA strand increases and polysomes are less likely to form. Despite these differences, CFPS can benefit from the relative slower synthesis rate and the distance between ribosomes by allowing nascent polypeptide chain more time and space to form desirable intra-peptide chain contacts, while decreasing the probability for undesirable, non-specific inter-peptide chain contacts, thereby increasing the probability of proper folding and decreasing the probability of aggregation[4]. As a result, from the CFPS system we can get more enzymes and with higher activity. |
| | + | </p><br/> |
| | | | |
| | + | <h3>d.Applications</h3> |
| | + | <p style="font-size:18px">The diversity of the cell-free systems allows in vitro synthesis of a wide range of proteins for a variety of downstream applications, such as screeening of enzymes activities. In the post-genomic era, cell-free protein synthesis has rapidly become the preferred approach for high-throughput functional and structural studies of proteins and a versatile tool for in vitro protein evolution and synthetic biology.</p> |
| | | | |
| − | <br/><br/><br/> <img src="https://static.igem.org/mediawiki/2016/4/4b/T--Tianjin--R-R_system5.png" alt="desktop">
| |
| − | <p style="font-size:15px">
| |
| − | Fig.4. Mechanism of TPA positive feedback system
| |
| − | </p>
| |
| − |
| |
| − | </div>
| |
| − | <div class="col-md-7">
| |
| | | | |
| − | <h3><b>3. TPA Positive Feedback Based Regulation System</b></h3>
| |
| − | <p style="font-size:18px"><br/>The next section is based on the TPA-inducing promoter. Considering the TPA degrading ability of <i>Rhodococcus jostii RHA1</i>, we believe there should be promoter that can sense and be induced by TPA. Luckily we find these three gene that have something to do with TPA degrading in <i>Rhodococcus jostii RHA1</i> can be induced significantly by TPA. The reason why these promoter can be induced by TPA is they have a leader sequence before the promoter sequence, we name it TPA inducible leader sequence (TILS). The gene of TPA transporting protein and regulation protein are also transformed into <i>Saccharomyces cerevisiae</i>. The TPA regulation protein is belong to the IclR family. This novel protein can combine the TILS and induce the expression of downstream gene when it combine the TPA molecule. Therefore, we insert the TILS before the enhanced promoter PGK1 so that we can make our promoter inducible by TPA.</p>
| |
| − |
| |
| − | </div>
| |
| − | </div>
| |
| − | <h2><b>Theoretical Background</b></h2>
| |
| − | <div class="row">
| |
| − | <div class="col-md-6">
| |
| − | <h3><b>1. The Cpx Regulation System<sup>[1]</sup></b></h3>
| |
| − | <br/> <p style="font-size:18px">In order to adapt to their changing environment,Escherichia coli bacterium need plenty of regulatory systems. The Cpx system is a three-component regulatory system which is kind of similar to the lactose operon.</p>
| |
| − | <p style="font-size:18px">The Cpx system consists of the histidine kinase CpxA, the response regulator CpxR and the periplasmic CpxP protein. CpxA is composed of a large periplasmic domain and a highly conserved cytosolic catalytic domain. Both domains are connected via two trans-membrane helices. CpxA has autophosphorylation, phosphor-transfer and phosphatase activities .Sensing envelope perturbation by an unknown feature, CpxA transmits a signal via a phosphorelay to CpxR, which in response acts as a transcription regulator of genes, whose products are mainly involved in envelope protein folding, detoxification and biofilm formation. The Cpx stress response is controlled by feedback inhibition CpxP acts at the initiation point of signal transduction by reducing CpxA auto-phosphorylation activity in the reconstituted CpxRA system.</p>
| |
| − | <p style="font-size:18px">The Cpx pathway is activated by a large number of different signals including elevated pH, increasing osmolarity, metals, altered membrane composition, overproduction of outer membrane lipoproteins and misfolded variants of maltose binding protein.</p>
| |
| − | <p style="font-size:18px">When the stress is at lower level, the CpxP protein combine with the CpxA to prevent CpxA from phosphorylating CpxR and when the stress changes at higher level, some signals lead to activation of the Cpx pathway. Particularly, misfolded protein such as MalE219 interacts directly with the periplasmic domain of CpxA, resulting in stimulation of CpxA phosphotransfer activity towards CpxR. </p>
| |
| | | | |
| | + | <h2><b>Experiment Design</b></h2> |
| | | | |
| − | </div>
| |
| − | <div class="col-md-6">
| |
| − | <br/><br/><br/><br/><br/>
| |
| − | <img src="https://static.igem.org/mediawiki/2016/9/9f/T--Tianjin--R-R_system9.png" alt="desktop">
| |
| − | <p style="font-size:15px"> Fig.5. The Cpx inclusion body responding system in <i>E.coli</i> <sup>[1]</sup></p>
| |
| − |
| |
| | | | |
| − |
| + | <p style="font-size:18px">Basically, we utilized the cell-free system to express the enzymes which had been modified in 22 different sites. Besides, we added a fluorescet protein, CFP, before the enzyme. And there is a flexible linker, GGGGSGGGGS , between them. So that we could detect the expression of enzymes by detecting expression of the fluorescent protein with a fluorescence readout instrument, for example, a microplate reader. We conceived that with this method we could acquire the best modifications by screening them in a high-throughput way. Then we used the proteins we got to degrade PET. </p> |
| − | </div>
| + | |
| − | </div>
| + | |
| | | | |
| | <div class="row"> | | <div class="row"> |
| − | <div class="col-md-5">
| + | <div class="col-md-2"></div> |
| − | | + | |
| − | | + | |
| − | | + | <div class="col-md-8"> |
| − | <br/><br/><br/><br/><br/><br/><img src="https://static.igem.org/mediawiki/2016/0/0d/T--Tianjin--R-R_system10.png" alt="desktop">
| + | <img src="https://static.igem.org/mediawiki/2016/c/ca/T--Tianjin--cf-e3.png" alt="desktop"> |
| | <p style="font-size:15px"> | | <p style="font-size:15px"> |
| − | Fig.6. The function of ddpX in <i>E.coli</i> under starvation conditions<sup>[2]</sup> | + | <br/> |
| | + | Fig.3.The expression vector in CFPS system |
| | </p> | | </p> |
| − |
| + | <div class="col-md-2"></div> |
| − | </div>
| + | |
| − | <div class="col-md-7">
| + | |
| | | | |
| − | <h3><b>2. DdpX cell lysis effect<sup>[2]</sup></b></h3><br/>
| |
| − | <p style="font-size:18px">DdpX, namely D-alanyl-D-alanine dipeptidase, is a kind of peptidoglycan hydrolase which can hydrolyze the D-Ala-D-Ala part in peptidoglycan molecule. As we all know, the cell wall of bacterial mainly consists of peptidoglycan, so the ddpX can hydrolyze the cell wall of bacterial.</p>
| |
| − | <p style="font-size:18px">It cannot be more strange that many bacterial own this kind of seemly dangerous gene in their genome. In fact, this gene also has many benefits to bacterial. In gram-positive bacterial, Vancomycin, a kind of antibiotics, can cause cell lysis because it can combine the D-Ala-D-Ala residue of peptidoglycan in cell wall and block the cross-linking of peptidoglycan. Some gram-positive bacterial like Enterococcus faecalis and Streptomyces toyocaensis have developed the resistance to the vancomycin because they have VanX gene, the homologue of ddpX gene, which can hydrolyze the D-Ala-D-Ala and transfer the D-Ala-D-Ala residue of peptidoglycan to D-Ala-D-Lac residue so that the vancomycin cannot combine the peptidoglycan.</p>
| |
| − | <p style="font-size:18px">However, in gram-negative bacterial like <i>E.coli</i>, which own the robust outer membrane that can resist the vancomycin, the hydrolase ddpX with the same effects also exists. This is strange because the gram-negative bacterial have no necessity to own this kind of seemly dangerous hydrolase. Actually the ddpX in <i>E.coli</i> has another vital use when they are under starvation conditions. The ddpX can hydrolyze the D-Ala-D-Ala in their cell wall to produce the D-Ala as the carbon source to maintain their life. This mechanism is only carried out when they are under starvation conditions. If the ddpX gene is overexpressed, the cell wall will be damaged and cell lysis will occur. </p>
| |
| | | | |
| | </div> | | </div> |
| − | </div>
| |
| − | <div class="row">
| |
| − | <div class="col-md-5">
| |
| − | <h3><b>3. TPA Positive Feedback Mechanism<sup>[3]</sup><sup>[4]</sup></b></h3>
| |
| − | <br/> <p style="font-size:18px">As we all know, PET is solid in normal condition. So it’s not easy for microorganisms to realize if there is any PET in the environment. For this reason, we designed the following regulating path.</p>
| |
| − | <p style="font-size:18px">We aim at finding a way to offer bacterial the ability to sense TPA so that it can produce more enzyme when TPA degraded by PETase exists in the environment.
| |
| − | We find the similar mechanism in the <i>Rhodococcus jostii RHA1</i>, which can make use of TPA as carbon source. We speculate that there must be the pathway we want in the <i>Rhodococcus jostii RHA1</i>. By the way, RHA1 is also well used in microbial consortia part of our project. In<i> Rhodococcus</i>, the distinct expression patterns of the TPA gene clusters indicate that they are independently regulated. The cluster contains gene encoding putative regulatory protein, namely tpaR. This gene encodes the regulatory protein of the IclR family, based on the presence of a conserved signature region. The regulator has helix-turn-helix domain and encodes regulator for its respective operons, which is consistent with the case for IclR-type regulatory proteins for other aromatic catabolism pathways. IclR-type positive regulators bind a sequence before their promoter DNA in the existence of inductor and start the transcription of downstream gene, so we need to express the regulator too.<sup>[4]</sup> Then the gene followed the promoter will be regulated by TPA. </p>
| |
| − |
| |
| − |
| |
| | | | |
| | </div> | | </div> |
| − | <div class="col-md-7">
| |
| − | <br/><br/><br/><br/><br/><br/><br/><br/><br/><br/><br/>
| |
| − | <img src="https://static.igem.org/mediawiki/2016/thumb/b/bd/T--Tianjin--R-R_system4.jpg/800px-T--Tianjin--R-R_system4.jpg" alt="desktop">
| |
| − | <p style="font-size:15px"> Fig.7. The TPA positive feedback effects found in <i>Rhodococcus jostii RHA1</i><sup>[3]</sup></p>
| |
| − | <br/><br/><br/>
| |
| − | <p style="font-size:18px">We find a promoter from the upstream of a gene named tpaAa regulated by TPA. It will express 300 times more when TPA exist. So we plan to transform the three genes into <i>Saccharomyces cerevisiae</i>. They respectively encode TPA transporter, TPA regulation protein and RFP bonded with the TILS. Then we can detect the intension of the red signal to measure the expression of the protein in distinct concentrations of TPA. </p>
| |
| − |
| |
| − |
| |
| − |
| |
| − |
| |
| − | </div>
| |
| − | </div>
| |
| − |
| |
| − | <h2><b>Experiment Design</b></h2>
| |
| | | | |
| | + | <p style="font-size:18px">How to characterize the degradation velocity is the main problem in our scheme. We analyzed the experiment consequences in two ways. For the first one, we rendered the enzymes degrade pNPa, a general substituent for the detection of PET. Then we measured the absorbance of pNP in the optical density of 400 nanometers, which is the degrading product of pNPa. For the second one, we detected the absorbance of MHET in the optical density of 260 nanometers, which is the product in the first step of PET degradation. |
| | + | </p> |
| | | | |
| | <div class="row"> | | <div class="row"> |
| − | <div class="col-md-5">
| + | <div class="col-md-2"></div> |
| − | <h3><b>1. Construction of Reporting System</b></h3>
| + | |
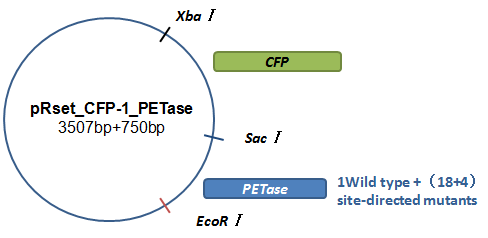
| − | <br/> <p style="font-size:18px">We use a common expression vector plasmid, pUC19, in <i>E.coli</i> to load our device, which consists of heterologous gene part (in this circumstance, PETase gene) and inclusion body reporting part. First of all, we transform the plasmid with part <i><i>BBa_K339007</i></i> from the kit shipped to us using the protocol in the instruction from iGEM official website. Then we use PCR to amplify this part with restriction endonuclease cutting sites <i>Xba1</i> and <i>Pst1</i> respectively on sense and anti-sense primers. Then we use corresponding restriction endonuclease to cut the part and plasmid pUC19 and then use T4 DNA ligase to link them together. The next step is to transform the PETase gene into the same plasmid. The initial gene synthetized does not has promoter and terminator so it cannot express. We have to cut the PETase gene and plasmid pET21A with <i>BamH1 </i> and Sal1 enzyme and link them together to transform the PETase gene into pET21A and then use PCR to amplify the T7 promoter-PETase gene-T7 terminator fragment added the restriction endonuclease cutting sites <i>EcoR1 </i> and Sac1. In this way, after we cut the recombinant plasmid pUC19 and T7 promoter-PETase gene-T7 terminator fragment with corresponding restriction endonucleases and link them together, we can obtain the complete device we want. </p> | + | |
| − | | + | <div class="col-md-8"> |
| − | | + | <img src="https://static.igem.org/mediawiki/2016/c/c5/T--Tianjin--cf-steps.jpg" alt="desktop"> |
| − | </div>
| + | |
| − | <div class="col-md-7">
| + | |
| − | <br/><br/><br/><br/><br/><br/><br/><br/>
| + | |
| − | <img src="https://static.igem.org/mediawiki/2016/thumb/4/4e/T--Tianjin--R-R_system6.jpg/800px-T--Tianjin--R-R_system6.jpg" alt="desktop">
| + | |
| − | <p style="font-size:15px"> Fig.8. The construction process of our reporting system</p>
| + | |
| − | | + | |
| − | </div>
| + | |
| − | </div>
| + | |
| − | <div class="col-md-12">
| + | |
| − | <h3><b>2. Verification of RFP in the part <i><i>BBa_K339007</i></i></b></h3>
| + | |
| − | <p style="font-size:18px">The verification of RFP is carried out by using PCR to amplify the RFP gene with restriction endonuclease cutting sites <i>Xba1</i> and Sac1 added and then cut the RFP and plasmid pET21a with corresponding restriction endonuclease. Then the cut fragments are linked together and transformed into <i>E.coli</i> to express. Then we can detect the red fluorescence.</p>
| + | |
| − | | + | |
| − | </div>
| + | |
| − | | + | |
| − | | + | |
| − | <div class="col-md-12">
| + | |
| − | <h3><b>3. Method of Red Fluorescence Assay</b></h3>
| + | |
| − | <p style="font-size:18px">The red fluorescence is detected by 96-well Microplate Reader. The excitation wavelength is set at 584nm and the emission wavelength is set at 607nm. Considering the RFP has an advantage that it can be directly observed by bare eyes, we also use centrifugation to precipitate the bacterial and observe the color of sediment. The red color can be observed if the RFP is expressed. All the experiment including the latter mentioned regulation system use this assay method.</p>
| + | |
| − | | + | |
| − | </div>
| + | |
| − | | + | |
| − | | + | |
| − | <div class="col-md-12">
| + | |
| − | <h3><b>4. Culture and Expression Condition of <i>E.coli</i> in this experiment</b></h3>
| + | |
| − | <p style="font-size:18px">Tradition culture medium LB (5g/L yeast extracts, 10g/L peptone, 10g/L NaCl) is also used by us. Because of the ampicillin resistance gene in the plasmid pUC19 and pET21A, ampicillin (100μg/mL) is added to screen for the correctly transformed bacterial. 5mL bacterial are cultured in test tube at 37℃ with 200rpm shaking speed. IPTG is added to induce the expression of PETase gene after 6 hours.</p>
| + | |
| − | | + | |
| − | </div>
| + | |
| − | | + | |
| − | | + | |
| − | <div class="row">
| + | |
| − | <div class="col-md-7">
| + | |
| − | | + | |
| − | | + | |
| − | | + | |
| − | <br/><br/><br/><br/><br/><br/><br/><br/><img src="https://static.igem.org/mediawiki/2016/thumb/b/bd/T--Tianjin--R-R_system7.jpg/800px-T--Tianjin--R-R_system7.jpg" alt="desktop">
| + | |
| | <p style="font-size:15px"> | | <p style="font-size:15px"> |
| − | Fig.9. The construction process of our cell lysis based regulation system | + | <br/> |
| | + | Fig.4.Steps for integrated expression and activity screening of enzymes |
| | </p> | | </p> |
| − |
| + | <div class="col-md-2"></div> |
| − | </div>
| + | |
| − | <div class="col-md-5">
| + | |
| − | | + | |
| − | <h3><b>5. Construction of Cell Lysis Based Regulation System</b></h3><br/>
| + | |
| − | <p style="font-size:18px">This system has a great similarity to the reporting system above. Therefore it is easy to construct because we only need to change the RFP gene to the ddpX gene. However, there is no restriction endonuclease cutting site between the CpxR and RFP gene sequence according to the part map from the iGEM official website, so we have to use PCR to amplify the CpxR promoter solely and add restriction endonuclease cutting sites <i>Xba1</i> and <i>BamH1 </i> respectively in both end. The ddpX gene is obtained from the <i>E.coli</i> genome using colony PCR and the <i>BamH1 </i> and <i>EcoR1 </i> restriction endonuclease cutting sites are added respectively to both end. Then the three fragments, CpxR promoter, ddpX gene, and cut plasmid pET21a are linked together. Then the whole part is amplified by PCR with <i>Xba1</i> and <i>Pst1</i> restriction endonuclease cutting sites added respectively to both end. This way, we can easily cut down the former CpxR-RFP fragment and add the new CpxR-ddpX fragment to the plasmid pUC19. </p>
| + | |
| | | | |
| | | | |
| | </div> | | </div> |
| − | </div>
| |
| − |
| |
| − |
| |
| − | <div class="col-md-12">
| |
| − | <h3><b>6. Verification of ddpX Gene Effect</b></h3>
| |
| − | <p style="font-size:18px">Just like the verification of RFP mentioned before, the verification of ddpX is carried out in the similar way. The pET21a plasmid is cut by <i>BamH1 </i> and <i>EcoR1 </i> instead of <i>Xba1</i> and <i>EcoR1 </i>, so that the ddpX can be linked to the cut plasmid pET21a solely. Then we can detect if the cell lysis occurs.</p>
| |
| − |
| |
| − | </div>
| |
| − |
| |
| − | <div class="col-md-12">
| |
| − | <h3><b>7. Method of Cell Lysis Assay</b></h3>
| |
| − | <p style="font-size:18px">Cell lysis can be reflected by the OD600 of culture medium. The lower the value of OD600 is than the wild type <i>E.coli</i> at the same condition, the stronger the cell lysis effect will be. The OD600 is detected by 96-well Microplate Reader. In order to know the OD600 value continuously, the detection process works through the time of bacterial growth and we will obtain the OD600-Growing time curve. </p>
| |
| − |
| |
| − | </div>
| |
| − |
| |
| − | <div class="col-md-12">
| |
| − | <h3><b>8. Chassis selection for TPA Positive Feedback Based Regulation System</b></h3>
| |
| − | <p style="font-size:18px">As the explanation before, the TPA positive feedback system is derived from the TPA degradation metabolic pathway in <i>Rhodococcus jostii RHA1</i>. Considering the difficulty of conducting gene-scale operation in this unusual organism, we directly synthetize all the gene including tpaK, tpaR, and TILS. At first we want to use <i>E.coli</i> to test this device because of the easy and familiar operation. However, in this situation, we have to transform at least 3 plasmids and this cannot be more difficult for <i>E.coli</i>. Therefore, we use another familiar organism, <i>Saccharomyces cerevisiae</i>, as the chassis. In the preliminary experiment, we successfully transform 3 plasmids into <i>Saccharomyces cerevisiae</i>. </p>
| |
| − |
| |
| − | </div>
| |
| − |
| |
| − |
| |
| − | <div class="row">
| |
| − | <div class="col-md-5">
| |
| − | <h3><b>9. Construction of TPA Positive Feedback Based Regulation System</b></h3>
| |
| − | <br/> <p style="font-size:18px">We use common plasmids of <i>Saccharomyces cerevisiae</i>, pRS413, pRS415 and pYES2, to respectively load the TPA transporting protein gene, TPA regulation protein gene and TPA induced RFP gene. First of all, we use PCR to amplify all of these fragments and add different restriction endonuclease cutting sites. Then we cut the plasmids with corresponding restriction endonucleases. Then these cut fragments are linked according to the designed order and transformed into <i>Saccharomyces cerevisiae</i>. We screen for the correctly transformed cell by using the Sc-Ura-Leu-His plate.</p>
| |
| − |
| |
| | | | |
| | </div> | | </div> |
| − | <div class="col-md-7">
| + | <h2><b>Reference</b></h2> |
| − | <br/><br/><br/><br/> | + | |
| − | <img src="https://static.igem.org/mediawiki/2016/thumb/a/a1/T--Tianjin--R-R_system8.jpg/800px-T--Tianjin--R-R_system8.jpg" alt="desktop">
| + | |
| − | <p style="font-size:15px"> Fig.10. The construction process of our TPA Positive Feedback Based regulation system</p>
| + | |
| | | | |
| − | </div>
| + | <p style="font-size:16px"><i>[1]Gabriel Rosenblum and Barry S. Cooperman. Engine out of the Chassis: Cell-Free Protein Synthesis and its Uses. FEBS Lett. 2014 January 21; 588(2): 261–268. doi:10.1016/j.febslet.2013.10.016.<br/><br/> |
| − | </div>
| + | [2]Aarthi Chandrasekaran and Anup K. Singh. One-Pot, Microscale Cell-Free Enzyme Expression and Screening. DOI 10.1007/978-1-62703-782-2 Springer New York Heidelberg Dordrecht London.<br/><br/> |
| − | | + | [3] C. Eric Hodgman, Michael C. Jewett. Cell-free synthetic biology: Thinking outside the cell. Metabolic Engineering 14 (2012) 261–269. doi:10.1016/j.ymben.2011.09.002<br/><br/> |
| − | | + | [4] Shaorong Chong. Overview of Cell-Free Protein Synthesis: Historic Landmarks, Commercial |
| − | <div class="col-md-12">
| + | Systems, and Expanding Applications. Current Protocols in Molecular Biology 16.30.1-16.30.11, October 2014 |
| − | <h3><b>10. Culture and Expression Condition of <i>Saccharomyces cerevisiae</i> in this experiment</b></h3>
| + | DOI: 10.1002/0471142727.mb1630s108 |
| − | <p style="font-size:18px">Traditional YPD culture medium (22g/L glucose, 20g/L peptone, 10g/L yeast extracts) is used by us. Sc-Ura-Leu-His culture medium (22g/L glucose, 6.7g/L yeast nitrogen base, 1.224g/L nutrient deficiency mixture without Ura, His, Leu and Trp, 5mg/L Trp) is used to screen for correctly transformed cell. All the cells are cultured in 5mL medium at 30℃ with shaking speed of 200rpm. To induce the expression of RFP, we add TPA with different concentration. We first make up TPA standard solution with TPA concentration of 5g/L. Then we respectively add 0, 1μL, 10μL, 100μL, 1mL standard solution to the culture medium. </p> | + | <br/><br/> |
| − | | + | |
| − | </div>
| + | |
| − | <div class="col-md-12">
| + | |
| − | <h2><b>Expected Results</b></h2>
| + | |
| − |
| + | |
| − | <p style="font-size:18px">PETase and MHETase are two key enzymes in our project. However, as heterologous proteins, the expression of these two enzymes face many problems just like expressing other heterologous proteins before including the formation of inclusion body, the lack of regulation pathway, etc. We design this R-R system in order to express the two enzymes visibly and regularly. </p>
| + | |
| − | | + | |
| − | <li><p style="font-size:18px">First, we hope to directly observe the expression condition of our enzyme by color, when the inclusion body form, which means the overexpressing, the red color can be observed. </p></li>
| + | |
| − | | + | |
| − | <li><p style="font-size:18px">Second, when inclusion body form, the normal way to solve this problem is to use lysozyme and ultrasonic to break the cell and purify the protein, which is complex and time-consuming. We expect the cell lysis will automatically occur when the inclusion body form by using ddpX gene. </p></li>
| + | |
| − | | + | |
| − | <li><p style="font-size:18px">Third, we expect the chassis organism can sense the existence of TPA, the hydrolyze product of PET and using TPA as the induction of PETase gene. Thus if the degradation process start, this process can be even enhanced until the PET is used up.</p></li>
| + | |
| − | | + | |
| − | <h2><b>References</b></h2>
| + | |
| − | <p style="font-size:16px"><i>[1]Physiologie der Mikroorganismen, Humboldt Universitat zu Berlin, Chausseestr. Misfolded maltose binding protein MalE219 induces the CpxRA envelope stress response by stimulating phosphoryl transfer from CpxA to CpxR. Research in Microbiology 160 (2009) 396-400.<br/><br/>
| + | |
| − | [2]Ivan A. D. Lwssard and Christopher T. Walsh. VanX, a bacterial D-alanyl-D-alanine dipeptidase: Resistance, immunity, or survival function? Proc. Natl. Acad. Sci. USA. Vol. 96, pp. 11028–11032, September, 1999.<br/><br/> | + | |
| − | [3]Hirofumi Hara, Lindsay D. Eltis, Julian E. Davies. Transcriptomic Analysis Reveals a Bifurcated Terephthalate | + | |
| − | Degradation Pathway in Rhodococcus sp. Strain RHA1. Journal of Bacteriology, Mar. 2007, 189(5), 1641–1647.<br/><br/>
| + | |
| − | [4]Molina-Henares, A. J., T. Krell, M. E. Guazzaroni, A. Segura, and J. L. Ramos. 2006. Members of the IclR family of bacterial transcriptional regulators function as activators and/or repressors. FEMS Microbiol. Rev. 30: 157–186.</i></p><br/><br/> | + | |
| − | | + | |
| − | </div></div>
| + | |
| | | | |
| | <!-- section end --> | | <!-- section end --> |
| Line 303: |
Line 156: |
| | <!-- section start --> | | <!-- section start --> |
| | <!-- ================ --> | | <!-- ================ --> |
| − |
| + | </div> |
| | + | </div> |
| | | | |
| | | | |
| Line 311: |
Line 165: |
| | | | |
| | {{:Team:Tianjin/Templates/Sponsor|}} | | {{:Team:Tianjin/Templates/Sponsor|}} |
| − | {{:Team:Tianjin/Templates/AddJS|:Team:Tianjin/Community/js/modernir.js}}
| |
| − | {{:Team:Tianjin/Templates/AddJS|:Team:Tianjin/Community/js/isotope.js}}
| |
| − | {{:Team:Tianjin/Templates/AddJS|:Team:Tianjin/Community/js/backstretch.js}}
| |
| − | {{:Team:Tianjin/Templates/AddJS|:Team:Tianjin/Community/js/appear.js}}
| |
| − | {{:Team:Tianjin/Templates/AddJS|:Team:Tianjin/Community/js/template.js}}
| |