Difference between revisions of "Team:Tianjin/Note/Consortium"
| Line 1: | Line 1: | ||
{{:Team:Tianjin/Templates/MaterialTheme|}} | {{:Team:Tianjin/Templates/MaterialTheme|}} | ||
| − | {{:Team:Tianjin/Templates/AddCSS|:Team:Tianjin/ | + | {{:Team:Tianjin/Templates/AddCSS|:Team:Tianjin/Experiment/R-R/style.css}} |
| − | {{:Team:Tianjin/Templates/AddCSS|:Team:Tianjin/ | + | {{:Team:Tianjin/Templates/AddCSS|:Team:Tianjin/Community/css/bootstrap.css}} |
| + | {{:Team:Tianjin/Templates/AddCSS|:Team:Tianjin/Community/css/font.css}} | ||
| + | {{:Team:Tianjin/Templates/AddCSS|:Team:Tianjin/Community/css/animation.css}} | ||
| + | {{:Team:Tianjin/Templates/AddCSS|:Team:Tianjin/Community/css/animate.css}} | ||
| − | <html | + | <html> |
| − | <head> | + | <head> |
| − | + | <meta charset="utf-8"> | |
| − | + | <title>Worthy</title> | |
| + | |||
| − | + | <!-- Mobile Meta --> | |
| + | <meta name="viewport" content="width=device-width, initial-scale=1.0"> | ||
| − | + | <!-- Favicon --> | |
| + | <link rel="shortcut icon" href="images/favicon.ico"> | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | </head> | |
| − | </head> | + | |
| + | <body class="no-trans"> | ||
| + | <!-- scrollToTop --> | ||
| + | <!-- ================ --> | ||
| + | <div class="scrollToTop"><i class="icon-up-open-big"></i></div> | ||
| − | + | <!-- header start --> | |
| − | <!-- | + | <!-- ================ --> |
| − | + | ||
| − | + | ||
| − | + | ||
| − | <!-- | + | |
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | <!-- header end --> | |
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| + | <!-- banner start --> | ||
| + | <!-- ================ --> | ||
| − | |||
| − | |||
| − | <!------------------- | + | <!-- section start --> |
| + | <!-- ================ --> | ||
| + | <div class="section clearfix object-non-visible" data-animation-effect="fadeIn"> | ||
| + | <div class="container"> | ||
| + | <div></br></div><div></br></div><div></br></div> | ||
| + | <!-- <div class="row"> --> | ||
| + | <div class="col-md-12"> | ||
| + | <h1 id="about" class="title text-center">Experiment of <span>Bacteria Consortium</span></h1> | ||
| + | <h2><b>Overview</b></h2> | ||
| + | <p style="font-size:18px">After Yoshida and his co-workers found and isolated <i>Ideonella sakaiensis 201-F6</i>, which produced two enzymes to degrades PET, we kept very high interests at their works and also came up with many ordinary ideas to increase the efficiency of degradation reaction. Bacteria consortium is one of the most creative ideas.<br/><br/> | ||
| + | The inspiration of this idea comes from nature and also learns from nature. Actually, bacteria never exist alone in our nature, they co-work and cooperate together to achieve an aim or live better in a special condition. Thinking from this point, we established a special bacteria consortium for this enzyme catalysis reaction. | ||
| + | </p> | ||
| + | |||
| + | <div class="row"> | ||
| + | <div class="col-md-8"> | ||
| + | <br/><br/><br/><br/> | ||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | + | <img src="https://static.igem.org/mediawiki/2016/thumb/2/2c/T--Tianjin--experiment-1.jpg/1043px-T--Tianjin--experiment-1.jpg" alt="desktop"> | |
| − | + | ||
| − | |||
| − | |||
| − | |||
| − | |||
| − | < | + | |
| − | < | + | <br/><p style="font-size:18px;text-align:center">Fig.1 Division of work in bacteria consortium</p> |
| + | <div class="space"></div> | ||
| + | </div> | ||
| + | <div class="col-md-4"> | ||
| + | <h3><b>1. Optimization of Culture Conditions</b><br/><br/> | ||
| + | <p style="font-size:18px">In order to improve efficiency of degrading PET, we are determined to co-culture <i> Pseudomonas putida KT2440, Rhodococcus jostii RHA1 </i>and <i>Bacillus stubtilis 168</i> (or <i> Bacillus stubtilis DB 104</i>). In our bacteria consortium, work of degradation is divided several parts as follows:<br/><br/> | ||
| + | 1.<i>Rhodococcus jostii RHA1 </i> is responsible for degrading TPA (terephthalic acid) to remove substrate inhibition;<br/><br/> | ||
| + | 2. <i> Pseudomonas putida KT2440</i> is responsible for degrading EG (ethylene glycol) to remove substrate inhibition, and contribute to produce degradable plastics PHA (polyhydroxyalkanoate).<br/><br/> | ||
| + | 3. <i>Bacillus stubtilis 168</i> (or <i> Bacillus stubtilis DB 104</i>) is responsible for secreting PETase and MHETase as the main player of degrading PET. | ||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
<br/> | <br/> | ||
| + | </p> | ||
| + | |||
| + | </div> | ||
| + | </div> | ||
| + | |||
| + | <div class="row"> | ||
| + | <div class="col-md-3"> | ||
| + | <p style="font-size:18px"> | ||
| + | Whereas, if our bacteria consortium want to achieve their aim, they must work in harmony, therefore, it is necessary find a appropriate environment where these bacteria can normally or supernormally work together. <br/><br/> | ||
| + | Primarily, we try several kinds of medium and decide to use W medium in the end; next, we optimize culture conditions by change carbon source, nitrogen source and some ions, then, we check growing situations and conditions of the degrading PET, TPA and EG; eventually, 1we can find out a suitable culture condition to co-culture our bacteria consortium. | ||
| − | |||
| − | |||
| − | |||
| − | |||
| − | < | + | </p> |
| − | <div | + | </div> |
| − | + | ||
| + | |||
| + | <div class="col-md-9"> | ||
| − | + | <img src="https://static.igem.org/mediawiki/2016/thumb/a/a4/T--Tianjin--experiment-2.jpg/1200px-T--Tianjin--experiment-2.jpg" alt="desktop"> | |
| − | + | ||
| − | + | ||
| − | < | + | <p style="font-size:18px;text-align:center"> |
| − | + | ||
| − | + | ||
<br/> | <br/> | ||
| + | Fig.2 Idea about optimization of culture conditions</div> | ||
| + | </p> | ||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | + | ||
| − | + | </div> | |
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
</div> | </div> | ||
| − | + | ||
| + | <div class="row"> | ||
| + | <div class="col-md-12"> | ||
| + | <h3><b>2. Modification of <i> Pseudomonas putida KT2440</i></b></h3> | ||
| + | <br/> <p style="font-size:18px"><i>P.putida KT2440</i> is one of bacteria which can utilize ethylene glycol (EG) at a high speed and meanwhile produces mcl-PHA. In 1988, Lageveen and his co-workers first found mcl-PHA in <i>P.putida KT2440</i>. And then, the metabolism of producing PHA in <i>P.putida KT2440</i> was reasearched, which found the gene AcoA was the key gene in the procedure. José Manuel Borrero-de Acuña and his co-workers improved the yield by 33% by overexpressing AcoA. | ||
| + | <br/><br/> | ||
| + | From Björn Mückschel’s works, Ethylene Glycol Metabolism by Pseudomonas putida was found. The key enzymes were identified by comparative proteomics. In P. putida JM37, tartronate semialdehyde synthase (Gcl), malate synthase (GlcB), and isocitrate lyase (AceA) were found to be induced in the presence of ethylene glycol or glyoxylic acid. Under the same conditions, strain KT2440 showed induction of AceA only. | ||
| + | <br/><br/> | ||
| + | From those studies, we decided to overexpress AcoA and AceA in <i>P.putida KT2440</i> to help utilize EG as energy source for its growth. | ||
| + | <br/><br/> | ||
| + | </p> | ||
| + | </div> | ||
| + | |||
| + | </div> | ||
| + | |||
| + | <div class="row"> | ||
| + | |||
| + | <div class="col-md-12"> | ||
| + | <h3><b>3. Modification of <i> Bacillus subtilis</i></b></h3> | ||
| + | <p style="font-size:18px"><br/>After some attempt in <i>E.coli</i> and yeast, we look for a new type of host cells- <i>B.subtilis</i> for more secretion. In our experiment, the genes encoding two enzymes are for the first time expressed in <i>S.cerevisiae</i>. Increased yields of PETase and MHETase enzymes are achieved when <i>B. subtilis</i> strains 168 and DB104 (deficient in two and three extracellular proteases, respectively<sup>[1]</sup>) were transformed with the recombinant plasmid with the help of the enhanced promoter-p43.</p> | ||
| + | |||
| + | </div> | ||
| + | </div> | ||
| − | + | <div class="row"> | |
| + | |||
| + | <div class="col-md-12"> | ||
| + | <h3><b>4. A Controllable Lipid Producer | ||
| + | </b></h3> | ||
| + | <p style="font-size:18px"><br/>Cyanobacteria are excellent organisms for biofuel production. We thus have selected Cyanobacterium <i>Synechocystis sp. PCC 6803</i> as the source of carbon in our mixed bacteria system. Our target is simply to make the cyanobacteria lyse at the appropriate time by transforming a plasmid contained three bacteriophage-derived lysis genes which were placed downstream of a nickel-inducible signal transduction system into the <i>Synechocystis 6803</i>.</p> | ||
| + | <br/> <p style="font-size:18px">In this part of our project, Cyanobacterium Synechocystis sp. PCC 6803 was selected as a model organism as the source of carbon in our mixed bacteria system. We simply to establish a cell wall disruption process which could make the cyanobacteria lyse at the appropriate time. </p> | ||
| + | </div> | ||
| + | </div> | ||
| + | <div class="row"> | ||
| − | + | <div class="col-md-12"> | |
| − | + | ||
| − | + | <h3><b>5. Gene Knockout of <i>Escherichia coli</i> | |
| + | </b></h3> | ||
| + | <p style="font-size:18px"><br/>In order to make S. cerevisiae and E.coli better survival, we need to change the energy source from glucose to xylose. When E. coli metabolizes xylose it excretes acetate, which is inhibitory to its own growth. S. cerevisiae cannot metabolize xylose but can use acetate as the sole carbon source without producing ethanol which is toxic to E.coli. For more acetate secretion, we successfully knocked off the atpF and atpH gene from the whole genome with the help of λ-red Recombination and I-SceI Cleavage. </p> | ||
| + | |||
| + | </div> | ||
| + | </div> | ||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | < | + | <h2><b>Theoretical Background</b></h2> |
| − | <div | + | <div class="row"> |
| − | + | <div class="col-md-12"> | |
| − | + | <h3><b>1. Degradation of Terephthalate</sup></b></h3> | |
| − | </ | + | <br/> <p style="font-size:18px"><i>Rhodococcus sp. strain RHA1</i> is thought to be capable of degrading a wide range of aromatic compounds including terephthalate acid (TPA). in 2006, a reliable pathway consisting of Distinct ring cleavage dioxygenase systems and protocatechuate (PCA) pathway was come up with, and the proposed degradation pathway for TPA is shown as below<sup>[2]</sup>.</p> |
| − | < | + | |
| − | + | ||
| − | + | ||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| + | </div></div> | ||
| − | + | <div class="row"> | |
| − | <div | + | <div class="col-md-12"> |
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | <div | + | |
| − | + | ||
<br/> | <br/> | ||
| − | </ | + | <div style="text-align:center"><img src="https://static.igem.org/mediawiki/2016/2/26/T--Tianjin--TPA_degratation.png" alt="desktop"></div> |
| + | <p style="font-size:18px;text-align:center">Fig.3 Proposed degradation pathway for TPA<sup>[2]</sup></p> | ||
| + | |||
| − | + | ||
| − | + | </div> | |
| − | + | </div> | |
| − | + | ||
| − | + | ||
| − | + | <div class="row"> | |
| − | <div | + | |
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | <div class="col-md-6"> | |
| − | <div | + | |
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | < | + | <br/><br/><br/><br/> |
| − | + | <div align="center"><img src="https://static.igem.org/mediawiki/2016/e/e0/T--Tianjin--EG_degratation.png" alt="desktop"></div> | |
| − | <div | + | |
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | </div> | |
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | <div class="col-md-6"> | |
| − | <div | + | |
| − | + | ||
| − | + | ||
| − | </div> | + | <h3><b>2. Degradation of Ethylene Glycol |
| − | <br/> | + | </b></h3><br/> |
| + | <p style="font-size:18px">By employing growth and bioconversion experiments, directed mutagenesis, and proteome analysis, it is found that <i>Pseudomonas putida KT2440</i> does not grow within 2 days of incubation, compared to <i>Pseudomonas putida JM37</i> which can grow rapidly under the same conditions. The key enzymes and specific differences between the two strains were identified by comparative proteomics. In <i>P. putida JM37</i>, tartronate semialdehyde synthase (Gcl), malate synthase (GlcB), and isocitrate lyase (AceA) were found to be induced in the presence of ethylene glycol or glyoxylic acid. Under the same conditions, strain KT2440 showed induction of AceA only. Postulated pathway for the metabolism of ethylene glycol in Pseudomonas putida strains KT2440 and JM37 is shown left<sup>[3]</sup>.</p> | ||
| + | <br/><br/><br/> | ||
| + | <p style="font-size:18px"> | ||
| + | Fig.4 Postulated pathway for the metabolism of ethylene glycol in <i>Pseudomonas putida </i>strains KT2440 and JM37. The enzymes and/or metabolites identified in response to ethylene glycol in KT2440 are depicted in black. Additional pathways identified in strain JM37 are shown in gray. Detailed descriptions of the pathways are given in the text.<sup>[3]</sup> | ||
| + | </p> | ||
| + | |||
| + | </div> | ||
| + | </div> | ||
| + | <div class="row"> | ||
| + | <div class="col-md-5"> | ||
| + | <h3><b>3. Production of Polyhydroxyalkanoate</b></h3> | ||
| + | <br/> <p style="font-size:18px"><i>Pseudomonas putida </i>is a natural producer of medium chain length polyhydroxyalkanoates (mcl-PHA), a polymeric precursor of bioplastics. A genome-based in silico model for <i>P. putida KT2440</i> metabolism was employed to identify potential genetic targets to be engineered for the improvement of mcl-PHA production using glucose as sole carbon source. Here, overproduction of pyruvate dehydrogenase subunit AcoA in the <i>P. putida KT2440</i> wild type led to an increase of PHA production. In controlled bioreactor batch fermentations PHA production was increased by 33% in the acoA overexpressing wild type in comparison to <i>P. putida KT2440</i>. Transcriptome analyses of engineered PHA producing P. putida in comparison to its parental strains revealed the induction of genes encoding glucose 6-phosphate dehydrogenase and pyruvate dehydrogenase. In addition, NADPH seems to be quantitatively consumed for efficient PHA synthesis, since a direct relationship between low levels of NADPH and high concentrations of the biopolymer were observed. In contrast, intracellular levels of NADH were found increased in PHA producing organisms. Central metabolism of <i>P. putida KT2440</i> is shown right<sup>[4]</sup>.</p> | ||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
</div> | </div> | ||
| − | <br/> | + | <div class="col-md-1"></div> |
| + | <div class="col-md-6"> | ||
| + | <br/><br/><br/><br/><br/> | ||
| + | <img src="https://static.igem.org/mediawiki/2016/thumb/2/2f/T--Tianjin--experiment-PHA.png/861px-T--Tianjin--experiment-PHA.png" alt="desktop"><br/> | ||
| + | <p style="font-size:18px;text-align:center">Fig.5 Central metabolism of <i>P. putida KT2440</i><sup>[4]</sup></p> | ||
| − | + | ||
| + | |||
| + | </div> | ||
| + | </div> | ||
| − | < | + | <div class="row"> |
| − | + | ||
| − | + | <div class="col-md-12"> | |
| − | <div | + | |
| − | + | ||
| − | + | ||
| + | <h3><b>4. Advantages of <i>B. subtilis</i> strains | ||
| + | </b></h3> | ||
| + | <p style="font-size:18px"><br/><i>Bacillus subtilis</i> has an excellent secretion ability, displays fast growth, and is a nonpathogenic bacterium free of endotoxin [5]. It can, therefore, be used in food, enzyme, and pharmaceutical industries and can replace <i>Escherichia coli</i> for protein expression. Furthermore, the extracellular heterogeneous proteins secreted from <i>B. subtilis</i> are more convenient for recovery and purification in large-scale production during downstream processing [6]. </p> | ||
| + | |||
| + | </div> | ||
| + | </div> | ||
| − | |||
| − | |||
| + | <div class="row"> | ||
| + | <div class="col-md-12"> | ||
| + | |||
| + | <h3><b>5. Enhanced promoter-p43 | ||
| + | </b></h3> | ||
| + | <p style="font-size:18px"><br/> In order to increase secretion, some enhanced promoters are necessary. However, native gene in a high-copy number plasmid was found to be unstable in <i>B. subtilis</i>[5]. To optimize the production and the stability of the expression vectors, both the promoter and the signal sequence of PETase were replaced by <i>B. subtilis</i> P43 promoter, a constitutively expressed promoter. This overcame the plasmid instability problem. </p> | ||
| + | |||
| + | </div> | ||
| + | </div> | ||
| − | + | <div class="row"> | |
| + | |||
| + | <div class="col-md-3"> | ||
| + | <br/><br/> | ||
| + | <div align="center"><img src="https://static.igem.org/mediawiki/2016/8/8d/T--Tianjin--experiment-3.png" alt="desktop"></div> | ||
| − | + | <p style="font-size:18px;text-align:center">Fig.6 construction of plasmid of cooperation of p43+psacB promoter<sup>[7]</sup> | |
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
</div> | </div> | ||
| − | <div | + | <div class="col-md-9"> |
| − | < | + | <h3><b>6. the cooperation of two promoters-p43+p<sub>sacB</sub> |
| − | < | + | </b></h3> |
| + | <p style="font-size:18px"><br/>Interestingly, the cooperation of two promoters in <i>B. subtilis</i> are easily found. For example, An endoglucanase from Bacillus akibai I-1 was successfully overexpressed in <i>Bacillus subtilis</i> 168 by the help of p43 promoter and the expression level of the recombinant enzyme was greatly enhanced by using the sucrose-inducible psacB promoter[7].The construction of plasmid is in the Figure 1 . Thus, we are willing to try whether the combination of p43 and psacB can make a difference in the secretion of enzyme.</p> | ||
| + | |||
| + | </div> | ||
| + | </div> | ||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| + | <div class="row"> | ||
| + | <div class="col-md-8"> | ||
| + | |||
| + | <h3><b>7. Lipid Producer | ||
| + | </b></h3> | ||
| + | <p style="font-size:18px"><br/> Photosynthetic microorganisms, including eukaryotic algae and cyanobacteria, are being optimized to overproduce numerous biofuel. According to previous data, algae accumulate large quantities of lipid as storage materials, but they do this when under stress and growing slowly. By contrast, cyanobacteria accumulate lipids in thylakoid membranes, which are associated with high levels of photosynthesis and a rapid growth rate. Thus, photo-synthetic bacteria have a natural advantage for producing lipids at a high rate. Furthermore, being prokaryotes can be improved by genetic manipulations much more readily than can eukaryotic algae. (Espaux L et al. 2015) Therefore, we decided to do something to make cyanobacteria ,the lipid producer, more appropriate for our project. | ||
| + | <br/><br/> | ||
| + | <i>Synechococcus elongatus PCC7942</i> has larger capacity of lipid production than <i>Synechocystis sp. PCC6803</i> but accumulates most of the product in the cell because of the imbalance of the rates of lipid production and secretion. Initially, we intended to do something to increase lipid secretion by knocking the <i>wzt</i> gene(Akihiro Kato et al. 2016), however, <i>Synechococcus elongatus PCC7942</i> wasn’t able to revive in two-week shaking cultivation. So we turned into <i>Synechocystis sp.PCC 6803</i>. </p> | ||
| + | |||
| + | </div> | ||
| + | <div class="col-md-4"> | ||
| + | <br/><br/><br/><br/><br/> | ||
| + | <div align="center"> <img src="https://static.igem.org/mediawiki/2016/9/9f/Igem-6803-e1.png" alt="desktop"></div> | ||
| + | <p style="font-size:18px;text-align:center">Fig.1. Lipid contents</p> | ||
| + | </div> | ||
| + | </div> | ||
| − | |||
| − | + | <div class="row"> | |
| − | < | + | <div class="col-md-6"> |
| − | < | + | <br/><br/><br/><br/><br/><br/><br/><br/><br/><br/> |
| − | < | + | <img src="https://static.igem.org/mediawiki/2016/d/d5/Igem-6803-e2.jpg" alt="desktop"> |
| − | < | + | |
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
<br/> | <br/> | ||
| + | <p style="font-size:18px;text-align:center">Fig.2. The functions of holin and endolysins in degrading the cell wall</p> | ||
| + | <br/><br/><br/> | ||
| + | </div> | ||
| + | |||
| + | <div class="col-md-6"> | ||
| + | <h3><b>8. Lipid Recovery From Biomass</b></h3> | ||
| + | <br/> <p style="font-size:18px">The first goal of our research was to facilitate lipid recovery from biomass. The scientific community widespread disrupts the cyanobacterial cell envelope to achieve the goal. (Seog JL et al. 1998)However, all these methods are not economical for large amounts of biomass or add additional cost and reduce the overall utility of the process. Our target is simply to make the cyanobacteria lyse at the appropriate time.</br> | ||
| + | We found that the cyanobacterial cell envelope is composed of 4 layers: the external surface layers ;the outer membrane; the polypeptidoglycan which is considerably thick, and the cytoplasmic membrane.( Hoiczyk E et al. 2000)To break up the peptidoglycan layer, we applied the holin-endolysin lysis strategy used by bacteriophages to exit bacterial cells(Wang IN et al. 2000). Endolysins are peptidoglycan-degrading enzymes that attack the covalent linkages of the peptidoglycans that maintain the integrity of the cell wall. In addition to endolysins, some auxiliary lysis factors are involved in cleaving the oligopeptide linkages between the peptidoglycan and the outer membrane lipoprotein. Holins are small membrane proteins that produce nonspecific lesions (holes) in the</p> | ||
| + | <br/> | ||
| + | <p style="font-size:18px"> cytoplasmic membrane from within, allow the endolysins and auxiliary lysis factors to gain access to the polypeptidoglycan layers, and trigger the lysis process. In this way, the cell wall is easy to break up.</p> | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | </div> | |
| − | + | </div> | |
| − | + | ||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | < | + | <div class="row"> |
| + | <div class="col-md-12"> | ||
| − | < | + | <h3><b>9. Control The Lysis System</b></h3> |
| − | < | + | <p style="font-size:18px"><br/>To control the appropriate time, a nickel sensing/responding signal system(Garcia-Dominguez M et al. 2000) was used to control the timing of the expression of phage lysis genes in Synechocystis 6803.</BR> |
| + | Our strategy for achieving our target is to construct a expression vector pCPC3031-Ni-13-19-15 introduced the Salmonella phage P22 lysis cassette (<I>13-19-15</I>) with a Ampicillin selection marker downstream of the promoter Pni, a nickel responding signal operon. Synechocystis 6803 with the pCPC3031-Ni-13-19-15 will lyse after Ni2+ addition. | ||
| + | </p> | ||
| − | + | ||
| − | + | </div> | |
| − | + | ||
| − | </div | + | </div> |
| − | + | ||
| − | < | + | <div class="row"> |
| − | <div | + | <div class="col-md-12"> |
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | < | + | <h3><b>10. Mixed bacteria vision</b></h3> |
| − | < | + | <p style="font-size:18px"><br/>In nature, microbes can form interacting communities to accomplish chemically difficult tasks through division of labor among different species. Hence, we want to mix <i>S. cerevisiae</i> and <i>E.coli</i> for two different usage[1]. The designed plasmids which have PETase and MHETase gene are put into <i>S. cerevisiae</i> for more secretion. In view of our R-R system, the inclusion body response switch(CpxR-ddpx-RFP) are put into <i>E.coli</i>. However, it isn’t easy to mix them together in glucose on account of inter-specific competition. So we use xylose as the only carbon source for further living. |
| − | + | </p> | |
| − | < | + | |
| − | < | + | |
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | < | + | |
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | </div> | |
| + | |||
| + | </div> | ||
| + | <div class="row"> | ||
| + | <div class="col-md-12"> | ||
| + | <h3><b>11. The mechanism of gene knockout</b></h3> | ||
| + | <p style="font-size:18px"><br/>we engineered <i>E. coli</i> to overproduce acetate by knocking out atpF and atpH gene from the whole E.coil genome and thereby further potentially improve the growth rate of <i>S. cerevisiae</i> by increasing the concentration of available substrate. We imitate λ-red Recombination system for genetic modification of the <i>Escherichia coli</i> chromosome, a three-resistance screening method involving λ-red recombination and I-SceI cleavage[2]. An intermediate strain is generated by integration of a resistance marker gene-tetracycline resistance genes and I-SceI recognition sites in or near the target gene locus, using λ-red PCR targeting. | ||
| + | </p> | ||
| + | <br/><br/> | ||
| − | + | </div> | |
| + | |||
| + | </div> | ||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | < | + | <h2><b>Experiment Design</b></h2> |
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | + | <li><h1 style="font-size:200%">Optimization of Culture Conditions</h1></li> | |
| − | < | + | |
| + | <div class="row"> | ||
| + | <div class="col-md-12"> | ||
| + | <h3><b>1. Find an Appropriate Medium</b></h3> | ||
| + | <br/> <p style="font-size:18px">Co-culture different pairs in improved W medium in the same condition (using two tubes in each group): </p> | ||
| + | <div align="center"> <img src="https://static.igem.org/mediawiki/2016/3/35/T--Tianjin--experiment-6.jpg" alt="desktop"></div> | ||
| + | <br/> <p style="font-size:18px">Co-culture different pairs in improved M9 medium in the same condition (using two tubes in each group):</p> | ||
| + | <div align="center"> <img src="https://static.igem.org/mediawiki/2016/3/35/T--Tianjin--experiment-6.jpg" alt="desktop"></div> | ||
| + | |||
| + | <br/> <p style="font-size:18px">Co-culture different pairs in LB medium in the same condition(using two tubes in each group):</p> | ||
| + | <div align="center"> <img src="https://static.igem.org/mediawiki/2016/3/35/T--Tianjin--experiment-6.jpg" alt="desktop"></div> | ||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| + | <br/> <p style="font-size:18px">Co-culture different pairs in YPD medium in the same condition (using two tubes in each group):</p> | ||
| + | <div align="center"> <img src="https://static.igem.org/mediawiki/2016/5/58/T--Tianjin--experiment-4.jpg" alt="desktop"></div> | ||
| + | <br/> <p style="font-size:18px"> | ||
| + | (PS: OD<sub>600</sub> of all the above bacteria solutions are 0.60, and these bacteria solutions are got by diluting all kinds of bacteria cultured in LB or YPD medium; and similarly hereinafter.)<br/><br/> | ||
| − | < | + | Culture all groups at 30℃ & 200 rpm for 3 days, check the bacterial concentration at OD<sub>600</sub> and observe these groups with microscope. |
| − | + | (P.S. We always extract 200μL samples to each well in 96-well plate, and similarly hereinafter.)<br/><br/> | |
| − | + | Eventually, we find W medium is most promising to co-culture these bacteria.<br/></p> | |
| − | + | ||
</div> | </div> | ||
| − | |||
| − | |||
| − | |||
</div> | </div> | ||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | + | <div class="col-md-12"> | |
| + | <h3><b>2. Nitrogen source</b></h3> | ||
| + | <p style="font-size:18px">We added some kinds of nitrogenous organic and inorganic compounds to W medium to improve initial W medium, and strategies of improvement are shown as follows.</p> | ||
| + | <div align="center"> <img src="https://static.igem.org/mediawiki/2016/0/07/T--Tianjin--experiment-5.jpg" alt="desktop"></div> | ||
| + | <br/> | ||
| + | <p style="font-size:18px">Add bacteria solution to media above as following table (use two tubes each group)</p> | ||
| + | <div align="center"> <img src="https://static.igem.org/mediawiki/2016/7/7f/T--Tianjin--table_8.22.png" alt="desktop"></div> | ||
| + | <p style="font-size:18px">Culture them at 30℃ & 200 rpm, checked the bacterial concentration at OD<sub>600</sub> and detect the concentration of TPA by UV at OD<sub>242</sub>, and then observe some samples with microscope.</p> | ||
| − | |||
| − | |||
| − | |||
| − | |||
| + | </div> | ||
| − | + | <div class="col-md-12"> | |
| − | + | <h3><b>3. Method of Red Fluorescence Assay</b></h3> | |
| − | + | <p style="font-size:18px">The red fluorescence is detected by 96-well Microplate Reader. The excitation wavelength is set at 584nm and the emission wavelength is set at 607nm. Considering the RFP has an advantage that it can be directly observed by bare eyes, we also use centrifugation to precipitate the bacterial and observe the color of sediment. The red color can be observed if the RFP is expressed. All the experiment including the latter mentioned regulation system use this assay method.</p> | |
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
</div> | </div> | ||
| − | |||
| − | < | + | <div class="col-md-12"> |
| − | < | + | <h3><b>4. Culture and Expression Condition of <i>E.coli</i> in this experiment</b></h3> |
| − | </ | + | <p style="font-size:18px">Tradition culture medium LB (5g/L yeast extracts, 10g/L peptone, 10g/L NaCl) is also used by us. Because of the ampicillin resistance gene in the plasmid pUC19 and pET21A, ampicillin (100μg/mL) is added to screen for the correctly transformed bacterial. 5mL bacterial are cultured in test tube at 37℃ with 200rpm shaking speed. IPTG is added to induce the expression of PETase gene after 6 hours.</p> |
| − | < | + | |
| − | + | ||
| − | < | + | |
| − | + | ||
</div> | </div> | ||
| − | |||
| − | |||
| − | |||
| − | |||
| − | + | <div class="col-md-7"> | |
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| + | <br/><br/><br/><br/><br/><br/><br/><br/><img src="https://static.igem.org/mediawiki/2016/thumb/b/bd/T--Tianjin--R-R_system7.jpg/800px-T--Tianjin--R-R_system7.jpg" alt="desktop"> | ||
| + | <p style="font-size:15px"> | ||
| + | Fig.9. The construction process of our cell lysis based regulation system | ||
| + | </p> | ||
| + | |||
| + | </div> | ||
| + | <div class="col-md-5"> | ||
| + | <h3><b>5. Construction of Cell Lysis Based Regulation System</b></h3><br/> | ||
| + | <p style="font-size:18px">This system has a great similarity to the reporting system above. Therefore it is easy to construct because we only need to change the RFP gene to the ddpX gene. However, there is no restriction endonuclease cutting site between the CpxR and RFP gene sequence according to the part map from the iGEM official website, so we have to use PCR to amplify the CpxR promoter solely and add restriction endonuclease cutting sites <i>Xba1</i> and <i>BamH1 </i> respectively in both end. The ddpX gene is obtained from the <i>E.coli</i> genome using colony PCR and the <i>BamH1 </i> and <i>EcoR1 </i> restriction endonuclease cutting sites are added respectively to both end. Then the three fragments, CpxR promoter, ddpX gene, and cut plasmid pET21a are linked together. Then the whole part is amplified by PCR with <i>Xba1</i> and <i>Pst1</i> restriction endonuclease cutting sites added respectively to both end. This way, we can easily cut down the former CpxR-RFP fragment and add the new CpxR-ddpX fragment to the plasmid pUC19. </p> | ||
| + | |||
| + | |||
| + | </div> | ||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | + | <div class="col-md-12"> | |
| − | + | <h3><b>6. Verification of ddpX Gene Effect</b></h3> | |
| + | <p style="font-size:18px">Just like the verification of RFP mentioned before, the verification of ddpX is carried out in the similar way. The pET21a plasmid is cut by <i>BamH1 </i> and <i>EcoR1 </i> instead of <i>Xba1</i> and <i>EcoR1 </i>, so that the ddpX can be linked to the cut plasmid pET21a solely. Then we can detect if the cell lysis occurs.</p> | ||
| + | </div> | ||
| − | < | + | <div class="col-md-12"> |
| + | <h3><b>7. Method of Cell Lysis Assay</b></h3> | ||
| + | <p style="font-size:18px">Cell lysis can be reflected by the OD600 of culture medium. The lower the value of OD600 is than the wild type <i>E.coli</i> at the same condition, the stronger the cell lysis effect will be. The OD600 is detected by 96-well Microplate Reader. In order to know the OD600 value continuously, the detection process works through the time of bacterial growth and we will obtain the OD600-Growing time curve. </p> | ||
| + | </div> | ||
| + | <div class="col-md-12"> | ||
| + | <h3><b>8. Chassis selection for TPA Positive Feedback Based Regulation System</b></h3> | ||
| + | <p style="font-size:18px">As the explanation before, the TPA positive feedback system is derived from the TPA degradation metabolic pathway in <i>Rhodococcus jostii RHA1</i>. Considering the difficulty of conducting gene-scale operation in this unusual organism, we directly synthetize all the gene including tpaK, tpaR, and TILS. At first we want to use <i>E.coli</i> to test this device because of the easy and familiar operation. However, in this situation, we have to transform at least 3 plasmids and this cannot be more difficult for <i>E.coli</i>. Therefore, we use another familiar organism, <i>Saccharomyces cerevisiae</i>, as the chassis. In the preliminary experiment, we successfully transform 3 plasmids into <i>Saccharomyces cerevisiae</i>. </p> | ||
| + | </div> | ||
| + | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | + | <div class="col-md-5"> | |
| + | <h3><b>9. Construction of TPA Positive Feedback Based Regulation System</b></h3> | ||
| + | <br/> <p style="font-size:18px">We use common plasmids of <i>Saccharomyces cerevisiae</i>, pRS413, pRS415 and pYES2, to respectively load the TPA transporting protein gene, TPA regulation protein gene and TPA induced RFP gene. First of all, we use PCR to amplify all of these fragments and add different restriction endonuclease cutting sites. Then we cut the plasmids with corresponding restriction endonucleases. Then these cut fragments are linked according to the designed order and transformed into <i>Saccharomyces cerevisiae</i>. We screen for the correctly transformed cell by using the Sc-Ura-Leu-His plate.</p> | ||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
</div> | </div> | ||
| − | <br/> | + | <div class="col-md-7"> |
| + | <br/><br/><br/><br/> | ||
| + | <img src="https://static.igem.org/mediawiki/2016/thumb/a/a1/T--Tianjin--R-R_system8.jpg/800px-T--Tianjin--R-R_system8.jpg" alt="desktop"> | ||
| + | <p style="font-size:15px"> Fig.10. The construction process of our TPA Positive Feedback Based regulation system</p> | ||
| + | </div> | ||
| + | </div> | ||
| + | <div class="col-md-12"> | ||
| + | <h3><b>10. Culture and Expression Condition of <i>Saccharomyces cerevisiae</i> in this experiment</b></h3> | ||
| + | <p style="font-size:18px">Traditional YPD culture medium (22g/L glucose, 20g/L peptone, 10g/L yeast extracts) is used by us. Sc-Ura-Leu-His culture medium (22g/L glucose, 6.7g/L yeast nitrogen base, 1.224g/L nutrient deficiency mixture without Ura, His, Leu and Trp, 5mg/L Trp) is used to screen for correctly transformed cell. All the cells are cultured in 5mL medium at 30℃ with shaking speed of 200rpm. To induce the expression of RFP, we add TPA with different concentration. We first make up TPA standard solution with TPA concentration of 5g/L. Then we respectively add 0, 1μL, 10μL, 100μL, 1mL standard solution to the culture medium. </p> | ||
| − | </ | + | </div> |
| + | <div class="col-md-12"> | ||
| + | <h2><b>Expected Results</b></h2> | ||
| + | |||
| + | <p style="font-size:18px">PETase and MHETase are two key enzymes in our project. However, as heterologous proteins, the expression of these two enzymes face many problems just like expressing other heterologous proteins before including the formation of inclusion body, the lack of regulation pathway, etc. We design this R-R system in order to express the two enzymes visibly and regularly. </p> | ||
| − | <li> | + | <li><p style="font-size:18px">First, we hope to directly observe the expression condition of our enzyme by color, when the inclusion body form, which means the overexpressing, the red color can be observed. </p></li> |
| − | < | + | |
| − | < | + | <li><p style="font-size:18px">Second, when inclusion body form, the normal way to solve this problem is to use lysozyme and ultrasonic to break the cell and purify the protein, which is complex and time-consuming. We expect the cell lysis will automatically occur when the inclusion body form by using ddpX gene. </p></li> |
| − | + | ||
| − | </ | + | <li><p style="font-size:18px">Third, we expect the chassis organism can sense the existence of TPA, the hydrolyze product of PET and using TPA as the induction of PETase gene. Thus if the degradation process start, this process can be even enhanced until the PET is used up.</p></li> |
| − | < | + | |
| − | <b>< | + | <h2><b>References</b></h2> |
| − | + | <p style="font-size:16px"><i>[1]Physiologie der Mikroorganismen, Humboldt Universitat zu Berlin, Chausseestr. Misfolded maltose binding protein MalE219 induces the CpxRA envelope stress response by stimulating phosphoryl transfer from CpxA to CpxR. Research in Microbiology 160 (2009) 396-400.<br/><br/> | |
| − | </ | + | [2]Ivan A. D. Lwssard and Christopher T. Walsh. VanX, a bacterial D-alanyl-D-alanine dipeptidase: Resistance, immunity, or survival function? Proc. Natl. Acad. Sci. USA. Vol. 96, pp. 11028–11032, September, 1999.<br/><br/> |
| − | <br/> | + | [3]Hirofumi Hara, Lindsay D. Eltis, Julian E. Davies. Transcriptomic Analysis Reveals a Bifurcated Terephthalate |
| + | Degradation Pathway in Rhodococcus sp. Strain RHA1. Journal of Bacteriology, Mar. 2007, 189(5), 1641–1647.<br/><br/> | ||
| + | [4]Molina-Henares, A. J., T. Krell, M. E. Guazzaroni, A. Segura, and J. L. Ramos. 2006. Members of the IclR family of bacterial transcriptional regulators function as activators and/or repressors. FEMS Microbiol. Rev. 30: 157–186.</i></p><br/><br/> | ||
| − | + | </div></div> | |
| − | + | ||
| − | </div | + | |
| − | + | ||
| − | + | <!-- section end --> | |
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| + | <!-- section start --> | ||
| + | <!-- ================ --> | ||
| + | <script type="text/javascript"> | ||
| + | var $VisualLightBoxParams$ = {autoPlay:true,borderSize:21,enableSlideshow:true,overlayOpacity:0.4,startZoom:true}; | ||
| + | </script> | ||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | < | + | </body> |
| − | < | + | </html> |
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| + | {{:Team:Tianjin/Templates/Sponsor|}} | ||
| − | + | {{:Team:Tianjin/Templates/AddJS|:Team:Tianjin/Experiment/lightbox.js}} | |
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | {{:Team:Tianjin/Templates/AddJS|:Team:Tianjin/ | + | |
| − | + | ||
Revision as of 12:48, 12 October 2016
Experiment of Bacteria Consortium
Overview
After Yoshida and his co-workers found and isolated Ideonella sakaiensis 201-F6, which produced two enzymes to degrades PET, we kept very high interests at their works and also came up with many ordinary ideas to increase the efficiency of degradation reaction. Bacteria consortium is one of the most creative ideas.
The inspiration of this idea comes from nature and also learns from nature. Actually, bacteria never exist alone in our nature, they co-work and cooperate together to achieve an aim or live better in a special condition. Thinking from this point, we established a special bacteria consortium for this enzyme catalysis reaction.

Fig.1 Division of work in bacteria consortium
1. Optimization of Culture Conditions
In order to improve efficiency of degrading PET, we are determined to co-culture Pseudomonas putida KT2440, Rhodococcus jostii RHA1 and Bacillus stubtilis 168 (or Bacillus stubtilis DB 104). In our bacteria consortium, work of degradation is divided several parts as follows:
1.Rhodococcus jostii RHA1 is responsible for degrading TPA (terephthalic acid) to remove substrate inhibition;
2. Pseudomonas putida KT2440 is responsible for degrading EG (ethylene glycol) to remove substrate inhibition, and contribute to produce degradable plastics PHA (polyhydroxyalkanoate).
3. Bacillus stubtilis 168 (or Bacillus stubtilis DB 104) is responsible for secreting PETase and MHETase as the main player of degrading PET.
Whereas, if our bacteria consortium want to achieve their aim, they must work in harmony, therefore, it is necessary find a appropriate environment where these bacteria can normally or supernormally work together.
Primarily, we try several kinds of medium and decide to use W medium in the end; next, we optimize culture conditions by change carbon source, nitrogen source and some ions, then, we check growing situations and conditions of the degrading PET, TPA and EG; eventually, 1we can find out a suitable culture condition to co-culture our bacteria consortium.

Fig.2 Idea about optimization of culture conditions
2. Modification of Pseudomonas putida KT2440
P.putida KT2440 is one of bacteria which can utilize ethylene glycol (EG) at a high speed and meanwhile produces mcl-PHA. In 1988, Lageveen and his co-workers first found mcl-PHA in P.putida KT2440. And then, the metabolism of producing PHA in P.putida KT2440 was reasearched, which found the gene AcoA was the key gene in the procedure. José Manuel Borrero-de Acuña and his co-workers improved the yield by 33% by overexpressing AcoA.
From Björn Mückschel’s works, Ethylene Glycol Metabolism by Pseudomonas putida was found. The key enzymes were identified by comparative proteomics. In P. putida JM37, tartronate semialdehyde synthase (Gcl), malate synthase (GlcB), and isocitrate lyase (AceA) were found to be induced in the presence of ethylene glycol or glyoxylic acid. Under the same conditions, strain KT2440 showed induction of AceA only.
From those studies, we decided to overexpress AcoA and AceA in P.putida KT2440 to help utilize EG as energy source for its growth.
3. Modification of Bacillus subtilis
After some attempt in E.coli and yeast, we look for a new type of host cells- B.subtilis for more secretion. In our experiment, the genes encoding two enzymes are for the first time expressed in S.cerevisiae. Increased yields of PETase and MHETase enzymes are achieved when B. subtilis strains 168 and DB104 (deficient in two and three extracellular proteases, respectively[1]) were transformed with the recombinant plasmid with the help of the enhanced promoter-p43.
4. A Controllable Lipid Producer
Cyanobacteria are excellent organisms for biofuel production. We thus have selected Cyanobacterium Synechocystis sp. PCC 6803 as the source of carbon in our mixed bacteria system. Our target is simply to make the cyanobacteria lyse at the appropriate time by transforming a plasmid contained three bacteriophage-derived lysis genes which were placed downstream of a nickel-inducible signal transduction system into the Synechocystis 6803.
In this part of our project, Cyanobacterium Synechocystis sp. PCC 6803 was selected as a model organism as the source of carbon in our mixed bacteria system. We simply to establish a cell wall disruption process which could make the cyanobacteria lyse at the appropriate time.
5. Gene Knockout of Escherichia coli
In order to make S. cerevisiae and E.coli better survival, we need to change the energy source from glucose to xylose. When E. coli metabolizes xylose it excretes acetate, which is inhibitory to its own growth. S. cerevisiae cannot metabolize xylose but can use acetate as the sole carbon source without producing ethanol which is toxic to E.coli. For more acetate secretion, we successfully knocked off the atpF and atpH gene from the whole genome with the help of λ-red Recombination and I-SceI Cleavage.
Theoretical Background
1. Degradation of Terephthalate
Rhodococcus sp. strain RHA1 is thought to be capable of degrading a wide range of aromatic compounds including terephthalate acid (TPA). in 2006, a reliable pathway consisting of Distinct ring cleavage dioxygenase systems and protocatechuate (PCA) pathway was come up with, and the proposed degradation pathway for TPA is shown as below[2].

Fig.3 Proposed degradation pathway for TPA[2]

2. Degradation of Ethylene Glycol
By employing growth and bioconversion experiments, directed mutagenesis, and proteome analysis, it is found that Pseudomonas putida KT2440 does not grow within 2 days of incubation, compared to Pseudomonas putida JM37 which can grow rapidly under the same conditions. The key enzymes and specific differences between the two strains were identified by comparative proteomics. In P. putida JM37, tartronate semialdehyde synthase (Gcl), malate synthase (GlcB), and isocitrate lyase (AceA) were found to be induced in the presence of ethylene glycol or glyoxylic acid. Under the same conditions, strain KT2440 showed induction of AceA only. Postulated pathway for the metabolism of ethylene glycol in Pseudomonas putida strains KT2440 and JM37 is shown left[3].
Fig.4 Postulated pathway for the metabolism of ethylene glycol in Pseudomonas putida strains KT2440 and JM37. The enzymes and/or metabolites identified in response to ethylene glycol in KT2440 are depicted in black. Additional pathways identified in strain JM37 are shown in gray. Detailed descriptions of the pathways are given in the text.[3]
3. Production of Polyhydroxyalkanoate
Pseudomonas putida is a natural producer of medium chain length polyhydroxyalkanoates (mcl-PHA), a polymeric precursor of bioplastics. A genome-based in silico model for P. putida KT2440 metabolism was employed to identify potential genetic targets to be engineered for the improvement of mcl-PHA production using glucose as sole carbon source. Here, overproduction of pyruvate dehydrogenase subunit AcoA in the P. putida KT2440 wild type led to an increase of PHA production. In controlled bioreactor batch fermentations PHA production was increased by 33% in the acoA overexpressing wild type in comparison to P. putida KT2440. Transcriptome analyses of engineered PHA producing P. putida in comparison to its parental strains revealed the induction of genes encoding glucose 6-phosphate dehydrogenase and pyruvate dehydrogenase. In addition, NADPH seems to be quantitatively consumed for efficient PHA synthesis, since a direct relationship between low levels of NADPH and high concentrations of the biopolymer were observed. In contrast, intracellular levels of NADH were found increased in PHA producing organisms. Central metabolism of P. putida KT2440 is shown right[4].

Fig.5 Central metabolism of P. putida KT2440[4]
4. Advantages of B. subtilis strains
Bacillus subtilis has an excellent secretion ability, displays fast growth, and is a nonpathogenic bacterium free of endotoxin [5]. It can, therefore, be used in food, enzyme, and pharmaceutical industries and can replace Escherichia coli for protein expression. Furthermore, the extracellular heterogeneous proteins secreted from B. subtilis are more convenient for recovery and purification in large-scale production during downstream processing [6].
5. Enhanced promoter-p43
In order to increase secretion, some enhanced promoters are necessary. However, native gene in a high-copy number plasmid was found to be unstable in B. subtilis[5]. To optimize the production and the stability of the expression vectors, both the promoter and the signal sequence of PETase were replaced by B. subtilis P43 promoter, a constitutively expressed promoter. This overcame the plasmid instability problem.
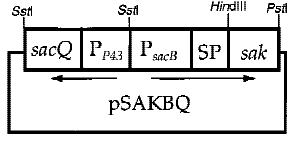
Fig.6 construction of plasmid of cooperation of p43+psacB promoter[7]
6. the cooperation of two promoters-p43+psacB
Interestingly, the cooperation of two promoters in B. subtilis are easily found. For example, An endoglucanase from Bacillus akibai I-1 was successfully overexpressed in Bacillus subtilis 168 by the help of p43 promoter and the expression level of the recombinant enzyme was greatly enhanced by using the sucrose-inducible psacB promoter[7].The construction of plasmid is in the Figure 1 . Thus, we are willing to try whether the combination of p43 and psacB can make a difference in the secretion of enzyme.
7. Lipid Producer
Photosynthetic microorganisms, including eukaryotic algae and cyanobacteria, are being optimized to overproduce numerous biofuel. According to previous data, algae accumulate large quantities of lipid as storage materials, but they do this when under stress and growing slowly. By contrast, cyanobacteria accumulate lipids in thylakoid membranes, which are associated with high levels of photosynthesis and a rapid growth rate. Thus, photo-synthetic bacteria have a natural advantage for producing lipids at a high rate. Furthermore, being prokaryotes can be improved by genetic manipulations much more readily than can eukaryotic algae. (Espaux L et al. 2015) Therefore, we decided to do something to make cyanobacteria ,the lipid producer, more appropriate for our project.
Synechococcus elongatus PCC7942 has larger capacity of lipid production than Synechocystis sp. PCC6803 but accumulates most of the product in the cell because of the imbalance of the rates of lipid production and secretion. Initially, we intended to do something to increase lipid secretion by knocking the wzt gene(Akihiro Kato et al. 2016), however, Synechococcus elongatus PCC7942 wasn’t able to revive in two-week shaking cultivation. So we turned into Synechocystis sp.PCC 6803.

Fig.1. Lipid contents

Fig.2. The functions of holin and endolysins in degrading the cell wall
8. Lipid Recovery From Biomass
The first goal of our research was to facilitate lipid recovery from biomass. The scientific community widespread disrupts the cyanobacterial cell envelope to achieve the goal. (Seog JL et al. 1998)However, all these methods are not economical for large amounts of biomass or add additional cost and reduce the overall utility of the process. Our target is simply to make the cyanobacteria lyse at the appropriate time. We found that the cyanobacterial cell envelope is composed of 4 layers: the external surface layers ;the outer membrane; the polypeptidoglycan which is considerably thick, and the cytoplasmic membrane.( Hoiczyk E et al. 2000)To break up the peptidoglycan layer, we applied the holin-endolysin lysis strategy used by bacteriophages to exit bacterial cells(Wang IN et al. 2000). Endolysins are peptidoglycan-degrading enzymes that attack the covalent linkages of the peptidoglycans that maintain the integrity of the cell wall. In addition to endolysins, some auxiliary lysis factors are involved in cleaving the oligopeptide linkages between the peptidoglycan and the outer membrane lipoprotein. Holins are small membrane proteins that produce nonspecific lesions (holes) in the
cytoplasmic membrane from within, allow the endolysins and auxiliary lysis factors to gain access to the polypeptidoglycan layers, and trigger the lysis process. In this way, the cell wall is easy to break up.
9. Control The Lysis System
To control the appropriate time, a nickel sensing/responding signal system(Garcia-Dominguez M et al. 2000) was used to control the timing of the expression of phage lysis genes in Synechocystis 6803.
Our strategy for achieving our target is to construct a expression vector pCPC3031-Ni-13-19-15 introduced the Salmonella phage P22 lysis cassette (13-19-15) with a Ampicillin selection marker downstream of the promoter Pni, a nickel responding signal operon. Synechocystis 6803 with the pCPC3031-Ni-13-19-15 will lyse after Ni2+ addition.
10. Mixed bacteria vision
In nature, microbes can form interacting communities to accomplish chemically difficult tasks through division of labor among different species. Hence, we want to mix S. cerevisiae and E.coli for two different usage[1]. The designed plasmids which have PETase and MHETase gene are put into S. cerevisiae for more secretion. In view of our R-R system, the inclusion body response switch(CpxR-ddpx-RFP) are put into E.coli. However, it isn’t easy to mix them together in glucose on account of inter-specific competition. So we use xylose as the only carbon source for further living.
11. The mechanism of gene knockout
we engineered E. coli to overproduce acetate by knocking out atpF and atpH gene from the whole E.coil genome and thereby further potentially improve the growth rate of S. cerevisiae by increasing the concentration of available substrate. We imitate λ-red Recombination system for genetic modification of the Escherichia coli chromosome, a three-resistance screening method involving λ-red recombination and I-SceI cleavage[2]. An intermediate strain is generated by integration of a resistance marker gene-tetracycline resistance genes and I-SceI recognition sites in or near the target gene locus, using λ-red PCR targeting.
Experiment Design
Optimization of Culture Conditions
1. Find an Appropriate Medium
Co-culture different pairs in improved W medium in the same condition (using two tubes in each group):

Co-culture different pairs in improved M9 medium in the same condition (using two tubes in each group):
Co-culture different pairs in LB medium in the same condition(using two tubes in each group):
Co-culture different pairs in YPD medium in the same condition (using two tubes in each group):

(PS: OD600 of all the above bacteria solutions are 0.60, and these bacteria solutions are got by diluting all kinds of bacteria cultured in LB or YPD medium; and similarly hereinafter.)
Culture all groups at 30℃ & 200 rpm for 3 days, check the bacterial concentration at OD600 and observe these groups with microscope.
(P.S. We always extract 200μL samples to each well in 96-well plate, and similarly hereinafter.)
Eventually, we find W medium is most promising to co-culture these bacteria.
2. Nitrogen source
We added some kinds of nitrogenous organic and inorganic compounds to W medium to improve initial W medium, and strategies of improvement are shown as follows.

Add bacteria solution to media above as following table (use two tubes each group)

Culture them at 30℃ & 200 rpm, checked the bacterial concentration at OD600 and detect the concentration of TPA by UV at OD242, and then observe some samples with microscope.
3. Method of Red Fluorescence Assay
The red fluorescence is detected by 96-well Microplate Reader. The excitation wavelength is set at 584nm and the emission wavelength is set at 607nm. Considering the RFP has an advantage that it can be directly observed by bare eyes, we also use centrifugation to precipitate the bacterial and observe the color of sediment. The red color can be observed if the RFP is expressed. All the experiment including the latter mentioned regulation system use this assay method.
4. Culture and Expression Condition of E.coli in this experiment
Tradition culture medium LB (5g/L yeast extracts, 10g/L peptone, 10g/L NaCl) is also used by us. Because of the ampicillin resistance gene in the plasmid pUC19 and pET21A, ampicillin (100μg/mL) is added to screen for the correctly transformed bacterial. 5mL bacterial are cultured in test tube at 37℃ with 200rpm shaking speed. IPTG is added to induce the expression of PETase gene after 6 hours.

Fig.9. The construction process of our cell lysis based regulation system
5. Construction of Cell Lysis Based Regulation System
This system has a great similarity to the reporting system above. Therefore it is easy to construct because we only need to change the RFP gene to the ddpX gene. However, there is no restriction endonuclease cutting site between the CpxR and RFP gene sequence according to the part map from the iGEM official website, so we have to use PCR to amplify the CpxR promoter solely and add restriction endonuclease cutting sites Xba1 and BamH1 respectively in both end. The ddpX gene is obtained from the E.coli genome using colony PCR and the BamH1 and EcoR1 restriction endonuclease cutting sites are added respectively to both end. Then the three fragments, CpxR promoter, ddpX gene, and cut plasmid pET21a are linked together. Then the whole part is amplified by PCR with Xba1 and Pst1 restriction endonuclease cutting sites added respectively to both end. This way, we can easily cut down the former CpxR-RFP fragment and add the new CpxR-ddpX fragment to the plasmid pUC19.
6. Verification of ddpX Gene Effect
Just like the verification of RFP mentioned before, the verification of ddpX is carried out in the similar way. The pET21a plasmid is cut by BamH1 and EcoR1 instead of Xba1 and EcoR1 , so that the ddpX can be linked to the cut plasmid pET21a solely. Then we can detect if the cell lysis occurs.
7. Method of Cell Lysis Assay
Cell lysis can be reflected by the OD600 of culture medium. The lower the value of OD600 is than the wild type E.coli at the same condition, the stronger the cell lysis effect will be. The OD600 is detected by 96-well Microplate Reader. In order to know the OD600 value continuously, the detection process works through the time of bacterial growth and we will obtain the OD600-Growing time curve.
8. Chassis selection for TPA Positive Feedback Based Regulation System
As the explanation before, the TPA positive feedback system is derived from the TPA degradation metabolic pathway in Rhodococcus jostii RHA1. Considering the difficulty of conducting gene-scale operation in this unusual organism, we directly synthetize all the gene including tpaK, tpaR, and TILS. At first we want to use E.coli to test this device because of the easy and familiar operation. However, in this situation, we have to transform at least 3 plasmids and this cannot be more difficult for E.coli. Therefore, we use another familiar organism, Saccharomyces cerevisiae, as the chassis. In the preliminary experiment, we successfully transform 3 plasmids into Saccharomyces cerevisiae.
9. Construction of TPA Positive Feedback Based Regulation System
We use common plasmids of Saccharomyces cerevisiae, pRS413, pRS415 and pYES2, to respectively load the TPA transporting protein gene, TPA regulation protein gene and TPA induced RFP gene. First of all, we use PCR to amplify all of these fragments and add different restriction endonuclease cutting sites. Then we cut the plasmids with corresponding restriction endonucleases. Then these cut fragments are linked according to the designed order and transformed into Saccharomyces cerevisiae. We screen for the correctly transformed cell by using the Sc-Ura-Leu-His plate.

Fig.10. The construction process of our TPA Positive Feedback Based regulation system
10. Culture and Expression Condition of Saccharomyces cerevisiae in this experiment
Traditional YPD culture medium (22g/L glucose, 20g/L peptone, 10g/L yeast extracts) is used by us. Sc-Ura-Leu-His culture medium (22g/L glucose, 6.7g/L yeast nitrogen base, 1.224g/L nutrient deficiency mixture without Ura, His, Leu and Trp, 5mg/L Trp) is used to screen for correctly transformed cell. All the cells are cultured in 5mL medium at 30℃ with shaking speed of 200rpm. To induce the expression of RFP, we add TPA with different concentration. We first make up TPA standard solution with TPA concentration of 5g/L. Then we respectively add 0, 1μL, 10μL, 100μL, 1mL standard solution to the culture medium.
Expected Results
PETase and MHETase are two key enzymes in our project. However, as heterologous proteins, the expression of these two enzymes face many problems just like expressing other heterologous proteins before including the formation of inclusion body, the lack of regulation pathway, etc. We design this R-R system in order to express the two enzymes visibly and regularly.
First, we hope to directly observe the expression condition of our enzyme by color, when the inclusion body form, which means the overexpressing, the red color can be observed.
Second, when inclusion body form, the normal way to solve this problem is to use lysozyme and ultrasonic to break the cell and purify the protein, which is complex and time-consuming. We expect the cell lysis will automatically occur when the inclusion body form by using ddpX gene.
Third, we expect the chassis organism can sense the existence of TPA, the hydrolyze product of PET and using TPA as the induction of PETase gene. Thus if the degradation process start, this process can be even enhanced until the PET is used up.
References
[1]Physiologie der Mikroorganismen, Humboldt Universitat zu Berlin, Chausseestr. Misfolded maltose binding protein MalE219 induces the CpxRA envelope stress response by stimulating phosphoryl transfer from CpxA to CpxR. Research in Microbiology 160 (2009) 396-400.
[2]Ivan A. D. Lwssard and Christopher T. Walsh. VanX, a bacterial D-alanyl-D-alanine dipeptidase: Resistance, immunity, or survival function? Proc. Natl. Acad. Sci. USA. Vol. 96, pp. 11028–11032, September, 1999.
[3]Hirofumi Hara, Lindsay D. Eltis, Julian E. Davies. Transcriptomic Analysis Reveals a Bifurcated Terephthalate
Degradation Pathway in Rhodococcus sp. Strain RHA1. Journal of Bacteriology, Mar. 2007, 189(5), 1641–1647.
[4]Molina-Henares, A. J., T. Krell, M. E. Guazzaroni, A. Segura, and J. L. Ramos. 2006. Members of the IclR family of bacterial transcriptional regulators function as activators and/or repressors. FEMS Microbiol. Rev. 30: 157–186.