Difference between revisions of "Team:Tianjin/Experiment/Protein Engineering"
ChenXinIGEM (Talk | contribs) |
ChenXinIGEM (Talk | contribs) |
||
| Line 407: | Line 407: | ||
<div class="col-md-2"></div> | <div class="col-md-2"></div> | ||
<div class="col-md-2 text-center"> | <div class="col-md-2 text-center"> | ||
| − | <a type="button" class="btn btn-next" href="https://2016.igem.org/Team:Tianjin/Note/Protein_Engineering"><img src="https://static.igem.org/mediawiki/2016/e/e7/T--Tianjin--button_note.png" width="120px"><p>See our Notes <br/>about | + | <a type="button" class="btn btn-next" href="https://2016.igem.org/Team:Tianjin/Note/Protein_Engineering"><img src="https://static.igem.org/mediawiki/2016/e/e7/T--Tianjin--button_note.png" width="120px"><p>See our Notes <br/>about Protein Modificiation</p></a> |
</div> | </div> | ||
<div class="col-md-2"></div> | <div class="col-md-2"></div> | ||
<div class="col-md-2 text-center"> | <div class="col-md-2 text-center"> | ||
| − | <a type="button" class="btn btn-next" href="https://2016.igem.org/Team:Tianjin/Demonstrate#proteinengineering"><img src="https://static.igem.org/mediawiki/2016/e/e1/T--Tianjin--button_result.png" width="120px"><p>See our Results</p></a> | + | <a type="button" class="btn btn-next" href="https://2016.igem.org/Team:Tianjin/Demonstrate#proteinengineering"><img src="https://static.igem.org/mediawiki/2016/e/e1/T--Tianjin--button_result.png" width="120px"><p>See our Results <br/> about Protein Engineering</p></a> |
</div> | </div> | ||
<div class="col-md-1"></div> | <div class="col-md-1"></div> | ||
Revision as of 17:56, 17 October 2016
Protein Engineering
Motivation
The degradation of PET is completed in two steps by PETase and MHETase. PETase hydrolyze PET to mono(2-hydroxyethyl) terephthalic acid (MHET), which will be further decomposed by MHETase into two monomers, terephthalic acid (TPA) and ethylene glycol (EG)[1] . In this two-step reaction system, MHET, the product of PETase-mediated hydrolysis of PET, was found to be a very minor component, which reveals rapid MHET metabolism[1], indicating the rate determining step in this reaction is the first step, hydrolysis of PET. So in order to accelerate the whole PET degradation speed, increasing the activity of PETase is rather crucial. To enhance PETase hydrolysis activity, we first tried to understand the mechanism of the hydrolysis reaction by generally confirming active sites of PETase and 3 dementional structure simulation.
Serine-based Catalytic Triad Mechanism & 3D Model Simulation
Since there is no x-ray structure for PETase , the mechanism of PETase hydrolysis activity can not be exactly identified. But the binding site and catalytic site can be generally inferred according to α/β hydrolase mechanism. Based on the efforts have been made to identify and characterize bacterial cutinases[2], α/β hydrolase fold family contain a highly conserved characteristic GXSXG motif. With sequence analysis, PETase was also found to contain an accordant GWSMG motif.
So we simulated a best fit model for PETase by SWISSMODEL, an automated comparative protein modeling server[3]. The template was Thc_Cut2, which shares 52% sequence identity with PETase[4]. As expected, the homology model of PETase displays a canonical α/β hydrolase fold with a Ser161-His237-Asp237 catalytic triad and a preformed oxyanion hole (Fig.1), suggesting a classic serine hydrolase mechanism.

Ribbon diagram of a predicted PETase model. The catalytic triad residues are shown as ball-and-sticks in green, formed by Ser161, His237 and Asp237, and the oxyanion hole binding site residues are in blue, formed by the main chain amides of Met161 and Tyr87.
Mutation Design Rationales
1.Hydrophobicity & space factor
Given that the hydrolysis of PET is a heterogeneous reaction, a prerequisite for its efficient reaction is an adequate interaction of the enzyme with the substrate at the solid-liquid interface. Since the substrate PET is hydrophobic polymer, both the hydrophobicity of the enzyme surface and the space around active site to accommodate PET may be the main factors for more efficient protein attachment and reaction activity[6]. In fact, tailoring enzymes for a more hydrophobic and larger substrate active site has been shown to facilitate PET hydrolysis by a fungal polyester hydrolase[5]. Also Silva et al. obtained a double mutant (Q132A/T101A) exhibiting a 2-fold increased hydrolysis activity against PET fibers by the substitution of spacious residues with alanine in its substrate binding site[6]. So we aimed to take the same strategy to modify hydrophobicity of the surface of PETase as well as broaden space near the active site to enhance the interaction between PETase and substrate.
2.Electrostatics factor
The electrostatic surface property in the vicinity to the active site was also found to be responsible for differences in hydrolysis efficiency. Exchange of the positively charged amino acids to the non-charged ones strongly increase the hydrolysis activity. In contrast, exchange of the uncharged amino acids by the negatively charged ones can lead to a complete loss of hydrolysis activity on PET films[7]. Changing the surface property is another strategy we took.
3.Conserved Residues
In another study, Wei and coworkers exchanged selected amino acid residues of TfCut2 involved in substrate binding with those in LC-cutinase(LCC) to relieve product inhibition and obtained enzyme variants with increased PET activity at 65℃ [8]. In the inspiration of their work as well as that the position of catalytic triad hold constant in different mature PET hydrolases with signal peptide excluded, we exchanged amino acid residues on the surface with highly conserved residues of the same position in other PET hydrolases, which can be a way to either increase the enzyme efficiency or confirm the essential sites which made PETase more efficient than other PET hydrolase.
Mutants
Based on the above rationales, our general strategies of mutation design are as follows:
1).According to the 3D structure of PETase we simulated, which shows the hydrophobicity of protein surface (Fig.2), we changed the hydrophilic amino acid residues on the surface with hydrophobic ones to enhance the surface hydrophobicity;
2). Modify the electrostatics of its surface by changing charged amino acid residues to the uncharged ones;
3). Based on 3D structure predicted and docking simulation with substrate, we found several spacious residues adjacent to active site and binding site, which may inhibit the interaction of protein and substrate due to its large body. We substituted them with smaller amino acids to expose the catalytic site and binding site;

The deeper the bule is, the more hydrophobic the protein surface is; On contrast, the deeper the brown is, the more hydrophilic the protein surface is.
4). We generated a multiple sequence alignment by ClustalX with seven serine hydrolases from NCBI conserved with GXSXG motif and catalytic triad and reported to be able to degrade PET (Table.1). Based on the multiple sequence alignment result(Fig.3), we exchanged some essential residues adjacent to active site and binding site with the conserved residues of the same position in the multiple sequences.

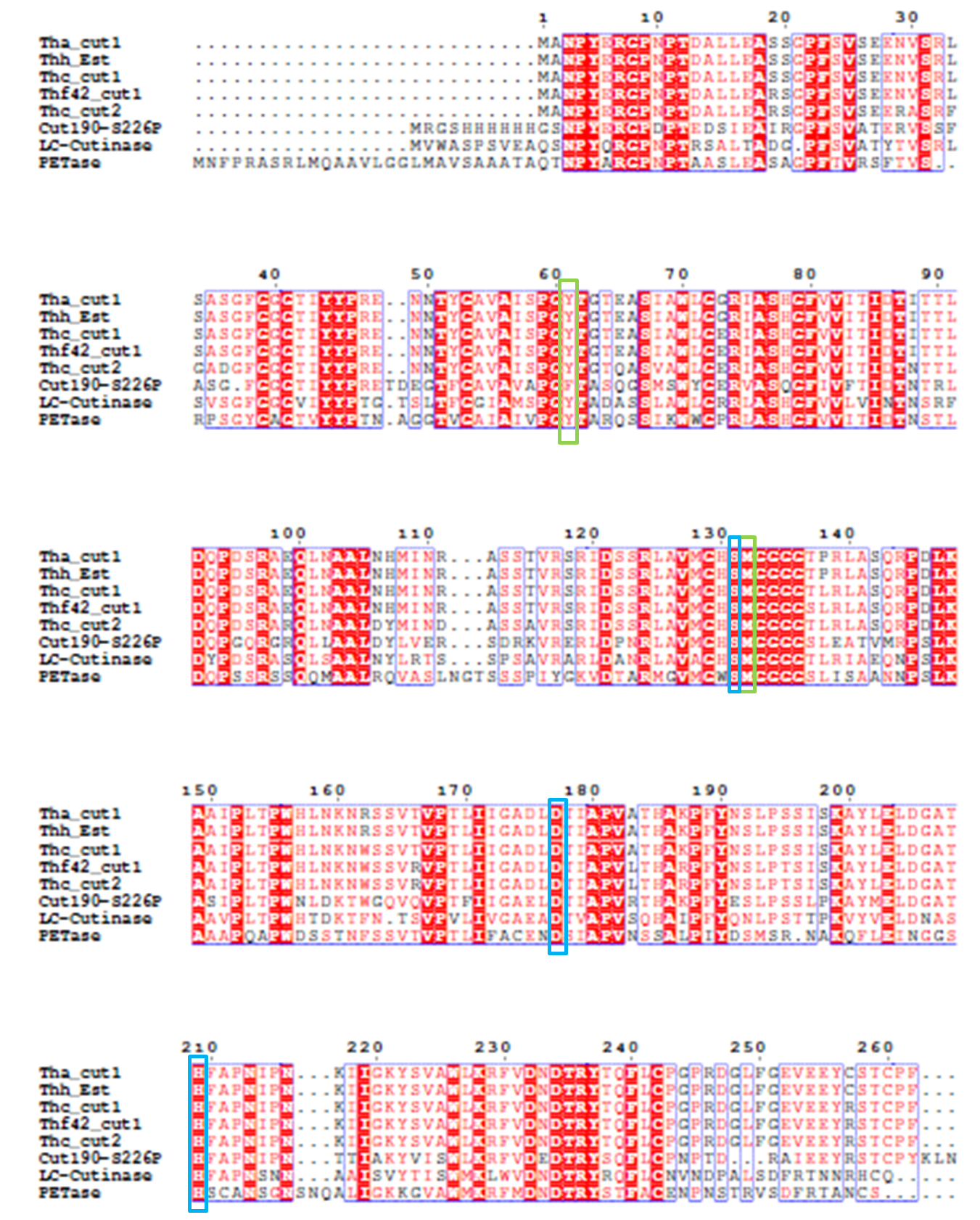
The catalytic triad is shaded in blue box and the binding site is shaded in green box.
The position of catalytic triad hold constant throughout the multiple sequences
in mature protein with signal peptide excluded.
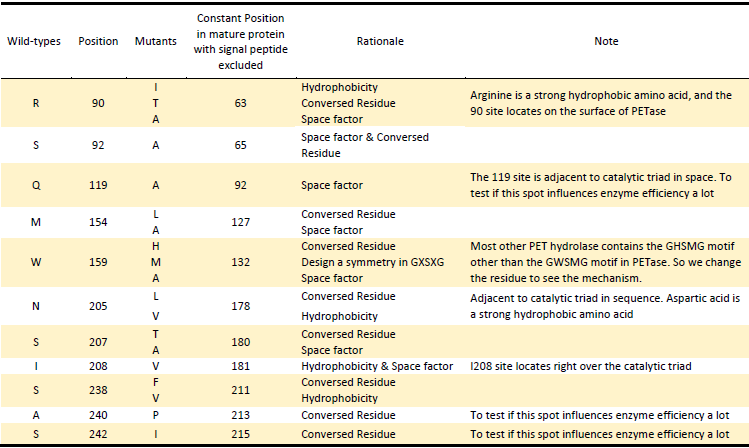
Based on the Rationales above, we designed 22 potential mutations
in purposes to enhance the efficiency of PETase hydrolysis activity
and confirm potential determining site of PETase hydrolysis.
Hight-throught Assay Strategy--CFPS
1.Overview
The CFPS system is the assay system in our project for the enzyme modification. The ability to produce a functional protein in the test tube, rather than in cells, is the essence of cell-free protein synthesis (CFPS)[11] .

The lack of high-throughput approaches for expression and screening of large enzyme libraries is a major bottleneck for current enzyme engineering efforts. To address this need, some researchers[12] have developed a high-throughput, fluorescence-based approach for rapid one-pot, microscale expression, and screening of different kinds of enzymes. To go further, we try to make our effert to achieve integration of cell-free protein expression with activity screening of enzymes( site-directed mutants of PETase).
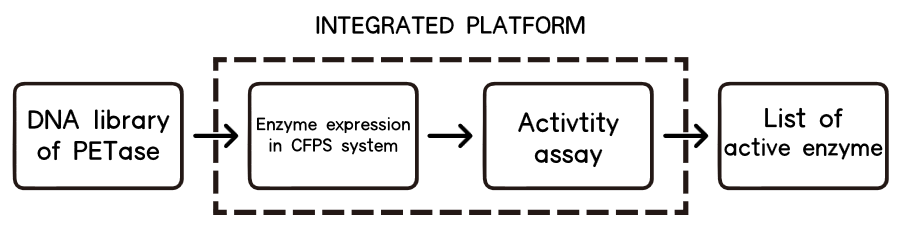
2.Background
a.Cell-free system plays an important role in synthetic biology
At its core, synthetic biology is inspired by the power and diversity of the living world. What is unique to synthetic biology is the application of an engineering-driven approach to accelerate the design-build-test loops required for reprogramming existing, and constructing new, biological systems. However, a major challenge exists because of our incomplete knowledge of how life works, the daunting complexity of cells, the unintended interference between native and synthetic parts, and (unlike typical engineered systems) the fact that cells evolve, have noise, and have their own agenda such as growth and adaptation[13].In addressing this questions, cell-free systems will play a very important role.
b.The system
Today cell-free protein synthesis(CFPS) has become a widely used method in synthetic biology as well as molecular biology. In addition to the challenges in vivo above, production of proteins using cell-free protein synthesis usually takes only a few hours, in contrast to production of proteins in cells, which typically takes days to weeks. In fact, even first-time users can often obtain newly synthesized proteins in one day using a commercial system.
c.Theoretic background
click here to find the CFPS Protocol
The compounds we will talk about are in the Feeding Buffer of the Protocol, which also plays the role of buffer solution.
For the transcription peorid, from tamplate DNA to mRNA, the necessties including 4 monosomes(adenosine monophosphate, guanosine monophosphate, cytidine monophosphate and uridine monophosphate, also be termed as AMP, GMP, CMP and UMP) of RNA, RNA polymersase, RNase inhibitor, energy resourse and other ions, such as Mg2+. Besides, our CFPS system including the lysate derived from Escherichia coli extract, which can provide sophisticated cofactors for the CFPS system.
The translation machinery is the engine of life. In order to synthesis the protein successfully, the 20 amino acids and ribosomes are obviously necessary. In E.coli CFPS the translation machinery is typically about 20-fold more dilute than in cell, decreasing the rates of initiation, elongation and protein accumulation . As well, the average distance between two adjacent ribosomes on a single mRNA strand increases and polysomes are less likely to form. Despite these differences, CFPS can benefit from the relative slower synthesis rate and the distance between ribosomes by allowing nascent polypeptide chain more time and space to form desirable intra-peptide chain contacts, while decreasing the probability for undesirable, non-specific inter-peptide chain contacts, thereby increasing the probability of proper folding and decreasing the probability of aggregation[14]. As a result, from the CFPS system we can get more enzymes and with higher activity.
d.Applications
The diversity of the cell-free systems allows in vitro synthesis of a wide range of proteins for a variety of downstream applications, such as screeening of enzymes activities. In the post-genomic era, cell-free protein synthesis has rapidly become the preferred approach for high-throughput functional and structural studies of proteins and a versatile tool for in vitro protein evolution and synthetic biology.
3.Experiment Design
Basically, we utilized the cell-free system to express the enzymes which had been modified in 22 different sites. Besides, we added a fluorescet protein, CFP, before the enzyme. And there is a flexible linker, GGGGSGGGGS , between them. So that we could detect the expression of enzymes by detecting expression of the fluorescent protein with a fluorescence readout instrument, for example, a microplate reader. We conceived that with this method we could acquire the best modifications by screening them in a high-throughput way. Then we used the proteins we got to degrade PET.
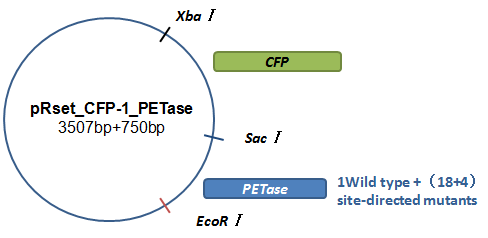
How to characterize the degradation velocity is the main problem in our scheme. We analyzed the experiment consequences in two ways. For the first one, we rendered the enzymes degrade pNPa, a general substituent for the detection of PET. Then we measured the absorbance of pNP in the optical density of 400 nanometers, which is the degrading product of pNPa. For the second one, we detected the absorbance of MHET in the optical density of 260 nanometers, which is the product in the first step of PET degradation.

Site-directed mutagenesis
Site-directed mutagenesis was performed using recombinant PCR technique[9]. Our modified approach is based on the PCR amplification of target PETase fragment by mutagenic primers divergently oriented but overlapping at their 5’ ends. The mutagenic nucleotides are located in both forward and reverse primers. The approach contained two rounds of PCR. In the PCR Round I, we use PETase fragment as template, respectively use original forward primer of PETase & mutagenic reverse primer, and original reverse primer & mutagenic forward primer as primers. The product of PCR Round I are portions of PETase gene with an overlapping region. In the PCR Round II, we use products of PCR Round I as two templates and original primers as primers to generate the final mutant fragment (Fig.4). And each specific mutation was verified by sequencing.

References
[1] Yoshida S, Hiraga K, Takehana T, et al. A bacterium that degrades and assimilates poly (ethylene terephthalate)[J]. Science, 2016, 351(6278): 1196-1199.
[2] Chen S, Tong X, Woodard R W, et al. Identification and characterization of bacterial cutinase[J]. Journal of Biological Chemistry, 2008, 283(38): 25854-25862.
[3] Schwede T, Kopp J, Guex N, Peitsch MC. SWISS-MODEL: an automated protein homologymodeling server. Nucleic Acids Research, 2003, 31 (13): 3381-3385
[4] Yoshida S, Hiraga K, Takehana T, et al. Supplementary Material For A bacterium that degrades and assimilates poly (ethylene terephthalate)
[5] Araújo R, Silva C, O’Neill A, et al. Tailoring cutinase activity towards polyethylene terephthalate and polyamide 6, 6 fibers[J]. Journal of Biotechnology, 2007, 128(4): 849-857.
[6] Silva C, Da S, Silva N, et al. Engineered Thermobifida fusca cutinase with increased activity on polyester substrates[J]. Biotechnology journal, 2011, 6(10): 1230-1239.
[7] Herrero Acero E, Ribitsch D, Dellacher A, et al. Surface engineering of a cutinase from Thermobifida cellulosilytica for improved polyester hydrolysis[J]. Biotechnology and bioengineering, 2013, 110(10): 2581-2590.
[8] Wei R, Oeser T, Schmidt J, et al. Engineered bacterial polyester hydrolases efficiently degrade polyethylene terephthalate due to relieved product inhibition[J]. Biotechnology and bioengineering, 2016.
[9] Ansaldi M, Lepelletier M, Méjean V. Site-specific mutagenesis by using an accurate recombinant polymerase chain reaction method [J]. Analytical biochemistry, 1996, 234(1): 110-111.
[10] Roth C, Wei R, Oeser T, et al. Structural and functional studies on a thermostable polyethylene terephthalate degrading hydrolase from Thermobifida fusca[J]. Applied microbiology and biotechnology, 2014, 98(18): 7815-7823.
[11]Gabriel Rosenblum and Barry S. Cooperman. Engine out of the Chassis: Cell-Free Protein Synthesis and its Uses. FEBS Lett. 2014 January 21; 588(2): 261–268. doi:10.1016/j.febslet.2013.10.016.
[12]Aarthi Chandrasekaran and Anup K. Singh. One-Pot, Microscale Cell-Free Enzyme Expression and Screening. DOI 10.1007/978-1-62703-782-2 Springer New York Heidelberg Dordrecht London.
[13] C. Eric Hodgman, Michael C. Jewett. Cell-free synthetic biology: Thinking outside the cell. Metabolic Engineering 14 (2012) 261–269. doi:10.1016/j.ymben.2011.09.002
[14] Shaorong Chong. Overview of Cell-Free Protein Synthesis: Historic Landmarks, Commercial
Systems, and Expanding Applications. Current Protocols in Molecular Biology 16.30.1-16.30.11, October 2014
DOI: 10.1002/0471142727.mb1630s108

