Difference between revisions of "Team:Tianjin/Experiment/Consortium"
ChenXinIGEM (Talk | contribs) |
|||
| Line 326: | Line 326: | ||
<div align="center"> | <div align="center"> | ||
<figure> | <figure> | ||
| − | <a href="https://static.igem.org/mediawiki/2016/d/d5/Igem-6803-e2.jpg" data-lightbox="no" data-title="Fig.8 The functions of holin and endolysins in degrading the cell wall"><img src="https://static.igem.org/mediawiki/2016/d/d5/Igem-6803-e2.jpg" ></a> | + | <a href="https://static.igem.org/mediawiki/2016/d/d5/Igem-6803-e2.jpg" data-lightbox="no" data-title="Fig.8 The functions of holin and endolysins in degrading the cell wall"><img class="img-responsive" src="https://static.igem.org/mediawiki/2016/d/d5/Igem-6803-e2.jpg" ></a> |
<figcation>Fig.8 The functions of holin and endolysins in degrading the cell wall</figcaption> | <figcation>Fig.8 The functions of holin and endolysins in degrading the cell wall</figcaption> | ||
</figure> | </figure> | ||
Revision as of 12:22, 18 October 2016
Bacteria Consortium
Overview
After Yoshida and his co-workers found and isolated Ideonella sakaiensis 201-F6, which produced two enzymes to degrades PET, we kept very high interests at their works and also came up with many ordinary ideas to increase the efficiency of degradation reaction. Bacteria consortium is one of the most creative ideas.
The inspiration of this idea comes from nature and also learns from nature. Actually, bacteria never exist alone in our nature, they co-work and cooperate together to achieve an aim or live better in a special condition. Thinking from this point, we established a special bacteria consortium for this enzyme catalysis reaction.

1. Optimization of Culture Conditions
In order to improve efficiency of degrading PET, we are determined to co-culture Pseudomonas putida KT2440, Rhodococcus jostii RHA1 and Bacillus stubtilis 168 (or Bacillus stubtilis DB 104). In our bacteria consortium, work of degradation is divided several parts as follows:
1.Rhodococcus jostii RHA1 is responsible for degrading TPA (terephthalic acid) to remove substrate inhibition;
2. Pseudomonas putida KT2440 is responsible for degrading EG (ethylene glycol) to remove substrate inhibition, and contribute to produce degradable plastics PHA (polyhydroxyalkanoate).
3. Bacillus stubtilis 168 (or Bacillus stubtilis DB 104) is responsible for secreting PETase and MHETase as the main player of degrading PET.
Whereas, if our bacteria consortium want to achieve their aim, they must work in harmony, therefore, it is necessary find a appropriate environment where these bacteria can normally or supernormally work together.
Primarily, we try several kinds of medium and decide to use W medium in the end; next, we optimize culture conditions by change carbon source, nitrogen source and some ions, then, we check growing situations and conditions of the degrading PET, TPA and EG; eventually, 1we can find out a suitable culture condition to co-culture our bacteria consortium.

2. Modification of Pseudomonas putida KT2440
P.putida KT2440 is one of bacteria which can utilize ethylene glycol (EG) at a high speed and meanwhile produces mcl-PHA. In 1988, Lageveen and his co-workers first found mcl-PHA in P.putida KT2440. And then, the metabolism of producing PHA in P.putida KT2440 was reasearched, which found the gene AcoA was the key gene in the procedure. José Manuel Borrero-de Acuña and his co-workers improved the yield by 33% by overexpressing AcoA.
From Björn Mückschel’s works, Ethylene Glycol Metabolism by Pseudomonas putida was found. The key enzymes were identified by comparative proteomics. In P. putida JM37, tartronate semialdehyde synthase (Gcl), malate synthase (GlcB), and isocitrate lyase (AceA) were found to be induced in the presence of ethylene glycol or glyoxylic acid. Under the same conditions, strain KT2440 showed induction of AceA only.
From those studies, we decided to overexpress AcoA and AceA in P.putida KT2440 to help utilize EG as energy source for its growth.
3. Modification of Bacillus subtilis
After some attempt in E.coli and yeast, we look for a new type of host cells- B.subtilis for more secretion. In our experiment, the genes encoding two enzymes are for the first time expressed in S.cerevisiae. Increased yields of PETase and MHETase enzymes are achieved when B. subtilis strains 168 and DB104 (deficient in two and three extracellular proteases, respectively[1]) were transformed with the recombinant plasmid with the help of the enhanced promoter-p43.
4. A Controllable Lipid Producer
Cyanobacteria are excellent organisms for biofuel production. We thus have selected Cyanobacterium Synechocystis sp. PCC 6803 as the source of carbon in our mixed bacteria system. Our target is simply to make the cyanobacteria lyse at the appropriate time by transforming a plasmid contained three bacteriophage-derived lysis genes which were placed downstream of a nickel-inducible signal transduction system into the Synechocystis 6803.
In this part of our project, Cyanobacterium Synechocystis sp. PCC 6803 was selected as a model organism as the source of carbon in our mixed bacteria system. We simply to establish a cell wall disruption process which could make the cyanobacteria lyse at the appropriate time.
Theoretical Background
1. Degradation of Terephthalate
Rhodococcus sp. strain RHA1 is thought to be capable of degrading a wide range of aromatic compounds including terephthalate acid (TPA). in 2006, a reliable pathway consisting of Distinct ring cleavage dioxygenase systems and protocatechuate (PCA) pathway was come up with, and the proposed degradation pathway for TPA is shown as below[2].


2. Degradation of Ethylene Glycol
By employing growth and bioconversion experiments, directed mutagenesis, and proteome analysis, it is found that Pseudomonas putida KT2440 does not grow within 2 days of incubation, compared to Pseudomonas putida JM37 which can grow rapidly under the same conditions. The key enzymes and specific differences between the two strains were identified by comparative proteomics. In P. putida JM37, tartronate semialdehyde synthase (Gcl), malate synthase (GlcB), and isocitrate lyase (AceA) were found to be induced in the presence of ethylene glycol or glyoxylic acid. Under the same conditions, strain KT2440 showed induction of AceA only. Postulated pathway for the metabolism of ethylene glycol in Pseudomonas putida strains KT2440 and JM37 is shown left[3].
3. Production of Polyhydroxyalkanoate
4. Advantages of B. subtilis strains
Bacillus subtilis has an excellent secretion ability, displays fast growth, and is a nonpathogenic bacterium free of endotoxin [5] . It can, therefore, be used in food, enzyme, and pharmaceutical industries and can replace Escherichia coli for protein expression. Furthermore, the extracellular heterogeneous proteins secreted from B. subtilis are more convenient for recovery and purification in large-scale production during downstream processing [6] .
5. Enhanced Promoter-p43
In order to increase secretion, some enhanced promoters are necessary. However, native gene in a high-copy number plasmid was found to be unstable in B. subtilis[5] . To optimize the production and the stability of the expression vectors, both the promoter and the signal sequence of PETase were replaced by B. subtilis P43 promoter, a constitutively expressed promoter. This overcame the plasmid instability problem.
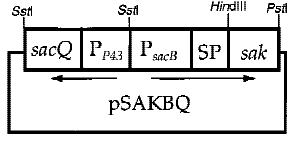
6. The Cooperation of Two Promoters-p43+psacB
Interestingly, the cooperation of two promoters in B. subtilis are easily found. For example, An endoglucanase from Bacillus akibai I-1 was successfully overexpressed in Bacillus subtilis 168 by the help of p43 promoter and the expression level of the recombinant enzyme was greatly enhanced by using the sucrose-inducible psacB promoter[7] .The construction of plasmid is in the Figure 1 . Thus, we are willing to try whether the combination of p43 and psacB can make a difference in the secretion of enzyme.
7. Lipid Producer
Photosynthetic microorganisms, including eukaryotic algae and cyanobacteria, are being optimized to overproduce numerous biofuel. According to previous data, algae accumulate large quantities of lipid as storage materials, but they do this when under stress and growing slowly. By contrast, cyanobacteria accumulate lipids in thylakoid membranes, which are associated with high levels of photosynthesis and a rapid growth rate. Thus, photo-synthetic bacteria have a natural advantage for producing lipids at a high rate. Furthermore, being prokaryotes can be improved by genetic manipulations much more readily than can eukaryotic algae. [8] Therefore, we decided to do something to make cyanobacteria ,the lipid producer, more appropriate for our project.
Synechococcus elongatus PCC7942 has larger capacity of lipid production than Synechocystis sp. PCC6803 but accumulates most of the product in the cell because of the imbalance of the rates of lipid production and secretion. Initially, we intended to do something to increase lipid secretion by knocking the wzt gene[9] (Akihiro Kato et al. 2016), however, Synechococcus elongatus PCC7942 wasn’t able to revive in two-week shaking cultivation. So we turned into Synechocystis sp.PCC 6803.


8. Lipid Recovery From Biomass
The first goal of our research was to facilitate lipid recovery from biomass. The scientific community widespread disrupts the cyanobacterial cell envelope to achieve the goal. [10] (Seog JL et al. 1998)However, all these methods are not economical for large amounts of biomass or add additional cost and reduce the overall utility of the process. Our target is simply to make the cyanobacteria lyse at the appropriate time. We found that the cyanobacterial cell envelope is composed of 4 layers: the external surface layers ;the outer membrane; the polypeptidoglycan which is considerably thick, and the cytoplasmic membrane. [11] ( Hoiczyk E et al. 2000)To break up the peptidoglycan layer, we applied the holin-endolysin lysis strategy used by bacteriophages to exit bacterial cells[12] (Wang IN et al. 2000). Endolysins are peptidoglycan-degrading enzymes that attack the covalent linkages of the peptidoglycans that maintain the integrity of the cell wall. In addition to endolysins, some auxiliary lysis factors are involved in cleaving the oligopeptide linkages between the peptidoglycan and the outer membrane lipoprotein. Holins are small membrane proteins that produce nonspecific lesions (holes) in the
cytoplasmic membrane from within, allow the endolysins and auxiliary lysis factors to gain access to the polypeptidoglycan layers, and trigger the lysis process. In this way, the cell wall is easy to break up.
9. Control The Lysis System
To control the appropriate time, a nickel sensing/responding signal system[13] (Garcia-Dominguez M et al. 2000) was used to control the timing of the expression of phage lysis genes in Synechocystis 6803.
Our strategy for achieving our target is to construct a expression vector pCPC3031-Ni-13-19-15 introduced the Salmonella phage P22 lysis cassette (13-19-15) with a Spectinomycin selection marker downstream of the promoter Pni, a nickel responding signal operon. Synechocystis 6803 with the pCPC3031-Ni-13-19-15 will lyse after Ni2+ addition.
Experiment Design
Optimization of Culture Conditions
1. Find an Appropriate Medium
Co-culture different pairs in improved W medium[17] in the same condition (using two tubes in each group):

Co-culture different pairs in improved M9 medium in the same condition (using two tubes in each group):
Co-culture different pairs in LB medium in the same condition(using two tubes in each group):
Co-culture different pairs in YPD medium in the same condition (using two tubes in each group):

(PS: OD600 of all the above bacteria solutions are 0.60, and these bacteria solutions are got by diluting all kinds of bacteria cultured in LB or YPD medium; and similarly hereinafter.)
Culture all groups at 30℃ & 200 rpm for 3 days, check the bacterial concentration at OD600 and observe these groups with microscope.
(P.S. We always extract 200μL samples to each well in 96-well plate, and similarly hereinafter.)
Eventually, we find W medium is most promising to co-culture these bacteria.
2. Nitrogen Source
We added some kinds of nitrogenous organic and inorganic compounds to W medium to improve initial W medium, and strategies of improvement are shown as follows.

Add bacteria solution to media above as following table (use two tubes each group)

Culture them at 30℃ & 200 rpm, checked the bacterial concentration at OD600 and detect the concentration of TPA by UV at OD242, and then observe some samples with microscope.
3. Carbon Source
We added some different kinds of carbonic organic compounds to W medium to improve initial W medium, and strategies of improvement are shown as follows.

Add bacteria solution to media above as following table (use two tubes each group)
Culture them at 30℃ & 200 rpm, checked the bacterial concentration at OD600 and detect the concentration of TPA by UV at OD242, and then observe some samples with microscope.
4. Mg2+ and Cl-
We add Mg2+ and Cl- to improve initial W medium, and strategies of improvement is shown as follows.

Add bacteria solution to media above as following table (use two tubes each group)
Culture them at 30℃ & 200 rpm, checked the bacterial concentration at OD600 and detect the concentration of TPA by UV at OD242, and then observe some samples with microscope.
5. Orthogonal Experiments
We added some different kinds of nitrogenous organic or inorganic compounds and carbonic organic compounds to W medium to improve initial W medium, and strategies of improvement are shown as follows.

(PS: Because concentration of TPA is lower actually, we decrease concentration of sodium terephthalate.)
Add bacteria solution to mediums above as following table (use two tubes each group)
Culture them at 30℃ & 200 rpm, checked the bacterial concentration at OD600 and detect the concentration of TPA by UV at OD242, and then observe some samples with microscope.
6. Temperture
Culture bacteria as following strategies at 37℃ & 200 rpm

Checked the bacterial concentration at OD600.
7. Preliminary Experiments for Degrading PET
We prepared media as following strategies:

(PS: Because concentration of TPA is lower actually, we decrease concentration of sodium terephthalate.)
Add bacteria solution to mediums above as following table (use two tubes each group)

Culture them at 30℃ & 200 rpm, checked the bacterial concentration at OD600 and detect the concentration of TPA by UV at OD242, and then observe some samples with microscope.
8. Degradation of PET
Culture bacteria as following strategies:

(PS: Later, we substitute Bacillus stubtilis DB 104 for Bacillus stubtilis 168.)
Culture them at 30℃ & 200 rpm, checked the bacterial concentration at OD600 and detect the concentration of TPA by UV at OD242, then, observe some sample with microscope.
We prepare some medium

(W medium + 3g/L KNO3 + 3g/L glucose + 3g/L sucrose + 2.5g/L sodium terephthalate), then we add 20μL Rhodococcus jostii RHA1, 0.5μL Pseudomonas putida KT2440, 10μL Bacillus stubtilis 168 (PETase) and 10μL Bacillus stubtilis DB104 (MHETase), after that, Culture them at 30℃ & 200 rpm, checked the bacterial concentration at OD600 and detect the concentration of TPA by UV at OD242, then, observe some sample with microscope.
9. Appendix
(1) We prepare a series of TPA standard solution and measure OD242, then plot standard curve.

(2)By dilution plating procedure[18], we find that concentration of Bacillus stubtilis 168 or
Bacillus stubtilis DB104 is approximately 0.5 * 10^8 when OD600=0.8; and Rhodococcus jostii RHA1 and Pseudomonas putida KT2440 are 1*10^8 and1.2*10^8 when OD600=0.8, because dilution plating procedure is relatively imprecise, these relationships between OD and concentration of bacteria are just for reference.
(3)Measure OD242
We firstly extract bacteria solutions to centrifugal tubes, then, centrifuge and extract supernatant 200μL to 96-well plate to measure OD242.
10. 16S rDNA
We validated our ideas that three kinds of bacteria were cooperating through the method of 16sRNA. The interesting thing is, three bacteria were living in harmony in modified W0 culture medium which changed the carbon source from glucose to sugar. we firstly tried to cultivate each of them(Rhodococcus RHA1, Pseudomonas putida KT2440, bacillus subtilis 168) in LB culture medium for amplification. Then we take a little of culture for next work.we design 2 primers in each bacteria which could amplify special 16s-DNA from whole genome.(Table 1)

Table.1 16S rDNA Primers Information

Table.2 The number of 16s-rDNA in the first colony-pcr : the provement of primers
We use orthogonal test to prove each bacteria had each special DNA stripe from the method of bacteria colony-pcr.In other words, we designed a 3*3 orthogonal test with the help of the bacteria liquid of three kinds and six primer sequences matched ribosome DNA of specific bacteria. The result was each bacteria had each special DNA stripe. Then we cultivate each of them and the mixture of three in W0 culture medium for amplification. Then we did the same 4*3(bacteria liquid and six primer sequences) orthogonal test. However, three bacteria failed to mix well, only rhodococcus lived.The number is collected in Table 2.
We cultivate each of them and the mixture of three in in modified W0 culture medium which changed the carbon source from glucose to sugar for amplification. Then we still did the same 4*3(bacteria liquid and six primer sequences) orthogonal test. Luckily, three bacteria mixed well after cultivating for 12 hours. The number is collected in Table 3.

Table.3 The number of 16s-rDNA in the second colony-pcr: the possibility of consortium
Modification of Pseudomonas putida KT2440
1. Overexpression of AcoA and AceA in P.putida KT2440
AcoA and AceA are the crucial genes for production of PHA and utilization of EG, so overexpressing these two genes is beneficial to improve the efficiency of degradation reaction and accumulation of PHA. Based on this idea, we established a expression vector which can be used in P.putida KT2440.
Based on the shuttle plasmid pBBR1MCS-2, we established the overexpression vector. The target genes were obtained by colony PCR and were ligated to the plasmid by T4 DNA ligase. The target genes are leaded by a T3 promoter.
After we established the vector, we transformed it to competent cell of E.coli and amplified it.

in P.putida KT2440
2. Electroporation of P.putida KT2440
Procedure:
(1) 10 ml LB + 0.1 ml Overnight culture of P.putida KT2440;
(2) Centrifugation at 5000 rpm for 10 min at 4 degrees centigrade;
(3) Wash the precipitation (P.putida KT2440) with ice cold Elec-Buffer * 3;
(4) Re-suspend in 20 microliter Elec-Buffer;
(5) 4 microliter or 9 microliter suspension + 1 microliter of DNA(50 ng);
(6) Place 5 microliter or 10 microliter samples in Electrode Pin(5.6 mm diameter?) (50 microliter in Electrode Pin(1 mm diameter)?);
(7) Electroporation at 1.2 kV, 200 Ω, and 25 μF.
(8) Add 1-2 ml SOC Medium and culture at 30 degrees centigrade for 2 h
(9) Spread on selection plate (LB agar with Kanamycin)
P.S. Elec-Buffer is 10% Glycerol.
3. Detection of PHA in P.putida KT2440
Procedure:
(1) take a certain amount of bacteria in the extraction bottle, add 15mL chloroform for each 1g bacteria, and place the extraction bottle in a high-pressure reaction kettle at 100 degrees Centigrade to react for 4h.
(2) After cooling, filter.
(3) Slowly add 10 times the volume of ethanol, cooling and stirring.
(4) Place at 4 degrees centigrade over night.
(5) After pressure reduction and pressure filtration, dehydrate the PHA in vacuum drying oven for 24h, weigh and record.
Modification of Bacillus subtilis
1. Recombinant Plasmid Construction and Transformation
E.coli with gene of PETase and MHETase and vector of pHP13-p43 has been inoculated in tubes of LB culture. The plasmid is constructed in advance(Figure 13).
The Plasmids were isolated and enzyme digested using EcoR I and BamH I restriction enzyme. After gel extraction to get right band, construction of pHP13-p43 + PETase and pHP13-p43 + MHETase were caught out. The successfully constructed vectors, pHP13-p43 + PETase and pHP13-p43 + MHETase, were respectively transferred into E.coli and then E.coli were cultivated on chloramphenicol-containing LB plates to filter positive colony.(Figure 14) The positive colony of PETase and of MHETase were inoculated into tubes containing LB with chloramphenicol. Then well-constructed vectors were isolated. Vectors were respectively transferred into B.subtilis and then B.subtilis were cultivated on erythromycin-containing LB plates to filter positive colony. Above all, we got 2 strains of B.subtilis respectively secret PETase and MHETase.


2. Bioactivity of PETase and MHETase
In order to test the bioactivity of PETase and MHETase, we have two methods. First, we use hydrolysis of pNPA through degradation rate. We cultivated wild B.subtilis and recombinant one in modified LB culture medium, which add PNPA solution through filtering(pNPA has a very low solubility in water).
After some days, we take 1 ml bacteria liquid each and centrifuge them for 12000r/min. Then we take the supernatant and measure solution absorbance at 400 nm, which is an obvious absorption peak of p-nitrophenol. If there is an an obvious absorption peak at 400 nm, we prove the bioactivity of PETase and MHETase in B.subtilis.
Second, we make orthogonal test. We cultivated wild B.subtilis and two recombinant one in LB culture medium(Figure 15). We put PET in wild one and one of the recombinant B.subtilis. Then, we imitate of the first methods and measure solution absorbance at 260 and 240 nm , which is an obvious absorption peak of MHET and TPA.

A Controllable Lipid Producer
1. Lysis genes(13-19-15)
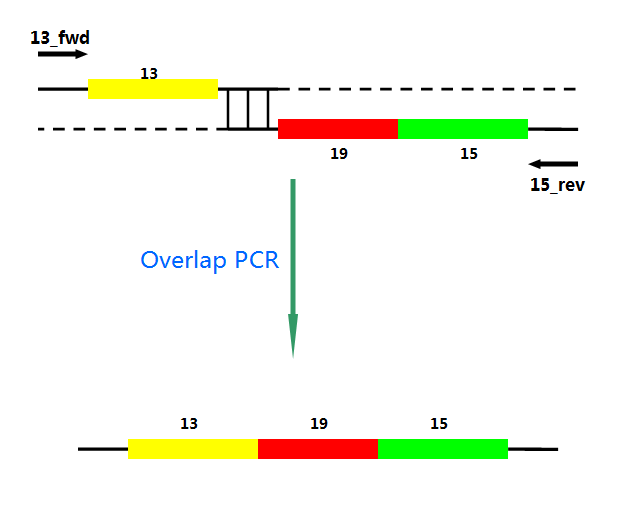
P22 gp13,P22 gp19 and P22 gp 15 are holins, endolysins and auxiliary lysis factors respectively. To construct the holin-endolysin lysis system, they should be connected together with defined sequence. First of all, we obtained the three lysis genes which were synthesized by GENEWIZ separately. Then we use PCR to amplify this part. TA cloning and ligation of Blunt-ended DNA on the T vector were our original idea.However, 19 and 15 were spliced via TA cloning according to our presumption. 13 and 19-15 were ligated PCR overlap extension method of Warrens et al.[19]( Warrens AN et al. 1997)

2.A Nickel Sensing/Responding Signal System(pCPC3031-Ni)
Ni activates the transcription of downstream genes of Pni and positively autoregulates its own synthesis. The amount of mRNA increased about 20-fold within 4 h after Ni addition. First, Pni was cloned into pCPC3031 and was amplified by using PCR. Then we cut the plasmid with Nru I, after that the lysis genes, 13-19-15, were placed downstream of Pni. What’ more, we handed in the plasimids for sequencing, which confirmed its correctness.
References
[1] He Wang, Ruijin Yang3,Xiao Hua, Wei Zhao, Wenbin Zhang. Functional Display of Active β-Galactosidase on Bacillus subtilis Spores Using Crust Proteins as Carriers
[2]Hirofumi Hara, Lindsay D. Eltis, Julian E. Davies, and William W. Mohn (2007): Transcriptomic Analysis Reveals a Bifurcated Terephthalate Degradation Pathway in Rhodococcus sp. Strain RHA1. In JOURNAL OF BACTERIOLOGY, Mar. 2007, p. 1641–1647. doi:10.1128/JB.01322-06
[3]Bjorn Mückschel,a Oliver Simon,b Janosch Klebensberger,a Nadja Graf,c Bettina Rosche,d Josef Altenbuchner,c Jens Pfannstiel,b Armin Huber,b and Bernhard Hauera (2012): Ethylene Glycol Metabolism by Pseudomonas putida. In Applied and Environmental Microbiology 78 (24), pp. 8531–8539.
[4]José Manuel Borrero-de Acuña, Agata Bielecka, Susanne Häussler, Max Schobert, Martina Jahn, Christoph Wittmann, Dieter Jahn1 and Ignacio Poblete-Castro (2014): Production of medium chain length polyhydroxyalkanoate in metabolic flux optimized Pseudomonas putida. In Microbial Cell Factories 13 (88).
[5] Sen-Lin Liu, Kun Du. Enhanced expression of an endoglucanase in Bacillus subtilis by using the sucrose-inducible sacB promoter and improved properties of the recombinant enzyme
[6] Ruiqiong Ye, June-Hyung Kim, Byung-Gee Kim, Steven Szarka, Elaine Sihota, Sui-Lam Wong High-Level Secretory Production of Intact, Biologically Active Staphylokinase from Bacillus subtilis
[7] Zhongjun Chen,Cai Heng ,Zhengying Li ,Xinle Liang , Shangguan Xinchen Expression and secretion of a single-chain sweet protein monellin in Bacillus subtilis by sacB promoter and signal peptide
[8]Espaux L, Mendez-Perez D, Li R, Keasling JD (2015) Synthetic biology for microbial production of lipid-based biofuels. Curr Opin Chem Biol. 29:58-65
[9]Akihiro Kato, Kazuhide Use, Nobuyuki Takatani, Kazutaka Ikeda, Miyuki Matsuura, Kouji Kojima (2016) Modulation of the balance of fatty acid production and secretion is crucial for enhancement of growth and productivity of the engineered mutant of the cyanobacterium Synechococcus elongates. Biotechnol Biofuels9:91-101.
[10]Seog JL, Byung-Dae Y, O. H-M (1998) Rapid method for the determination of lipid fromthe green alga Botryococcus braunii. Biotechnol Tech 12:553–556.
[11]Hoiczyk E, HanselA(2000) Cyanobacterial cell walls: News from an unusual prokaryotic envelope. J Bacteriol 182:1191–1199.
[12]Wang IN, Smith DL, Young R (2000) Holins: The protein clocks of bacteriophage infections. Annu Rev Microbiol 54:799–825.
[13]Garcia-Dominguez M, Lopez-Maury L, Florencio FJ, Reyes JC (2000) A gene clusterinvolved in metal homeostasis in the cyanobacterium Synechocystis sp. strain PCC 6803. J Bacteriol 182:1507–1514.
[14] Kang Zhou, Kangjian Qiao, Steven Edgar & Gregory Stephanopoulos.Distributing a metabolic pathway among a microbial consortium enhances production of natural products. doi:10.1038/nbt.3095
[15] Junjie Yang,Bingbing Sun,He Huang,Biao Chen,Chongmao Xu,Xin Wang,Jinle Liu ,Liuyang Diao .Multiple-site genetic modifications in Escherichia coli using lambda-Red recombination and I-SceI cleavage. doi:10.1007/s10529-015-1878-1
[16] T. B. Causey, S. Zhou, K. T. Shanmugam, and L. O. Ingram.Engineering the metabolism of Escherichia coli W3110 for the conversion of sugar to redox-neutral and oxidized products: Homoacetate production.
[17]MASASHI SETO, KAZUHIDE KIMBARA, MINORU SHIMURA, TAKASHI HATTA, MASAO FUKUDA, AND KEIJI YANO (1995): A Novel Transformation of Polychlorinated Biphenyls by Rhodococcus sp. Strain RHA1. In APPLIED AND ENVIRONMENTAL MICROBIOLOGY 61 (9), p. 3353–3358.
[18]Qian Ma (2015): The proteomic and metabolomic analyses of the consortia for vitamin C fermentation. Tianjin university.
[19]Warrens AN, Jones MD, Lechler RI (1997) Splicing by overlap extension by PCR using asymmetric amplification: An improved technique for the generation of hybrid proteins of immunological interest. Gene 186:29–35.

