Team:Tianjin/Safety
Proof
Our project mainly consists of three integrated sections--Protein Modification, Microbial Consortium and Reporting-Regulation System. In each section there are many concepts related to our experiment design. Our project focused on proving these concepts by constructing new parts. Our proof of concepts are divided into three sections respectively belong to the three sections of our whole project. The detailed proof method and relative constructed devices are as follows.
Protein Engineering
Outline
The real goal of the protein engineering part in our project was to create a more active form of PETase protein. We designed and expressed 22 mutations in cell-free protein synthesis system, a brand new way to do the high-throughout assay. And from the 22 mutations, we successfully screened out a mutation I208V with hydrolysis activity improved by two-fold compared to the wild-type PETase.
Rational Design
Serine-based Catalytic Triad Mechanism & 3D Model Simulation
Since PETase was found to contain a GWSMG motif in accordance with the GXGXG motif, which is characteristic and highly conserved in α/β hydrolase fold family, we simulated a best fit model for PETase by SWISSMODEL with a template as Thc_Cut2. As expected, the homology model of PETase displays a canonical α/β hydrolase fold with a Ser161-His237-Asp237 catalytic triad and a preformed oxyanion hole (Fig.1), suggesting a classic serine hydrolase mechanism.
Mutation Design
Based on the simulated 3D structure, we found the 208th (with signal peptide excluded) amino acid residue isoleucine situates right over the catalytic triad(Fig.2). It is apperant that the interaction of catalytic triad with substrate would be inhibited due to the space oppcupied by 208th isoleucine. So we decided to substitue 208th isoleucine with another smaller amino acid.
However, as the isoleucine is the most hydrophobic amino acid, it takes risk to substitue it with another one, as it may lead to the hydrophobic amino acid decreasing. On balance, we chose valine as substitution, for its hydrophobicity is quite close to isoleucine, and the volume of valine is much smaller than that of isoleucine.
The 3D structure of mutant I208V is as Fig.3. Comparing to wild-type PETase(Fig.2), the mutant I208V exposes the catalytic triad much more widely. And the assay shows the stratety worked well for improving PET hydrolysis activity. The results are as follows.
Plasmid construction and expression in CFPS system
Ligaion of digested pRset_CFP-1, digested CFP gene and digested PETase(I208V) gene in accordance with the 1:5:5 molecular ratio. The newly constructed plasmid is called pRset_CFP-1_PETase-I208V.Then, the plasmids pRset_CFP-1_PETase-I208V was put into the CFPS(Cell-Free Protein Synthesis) to synthesis the enzymes we expected.
Fig.1. Plasmid construction and expression in CFPS ststem.
Degradation data
After the enzymes were expressed in the system successfully, we used the proteins we got to degrade PET and detected the degradation product, MHET, which has no other characteristic adsorption peak except at 260nm.
Fig.2.Multispectral scanning for the degradation product.
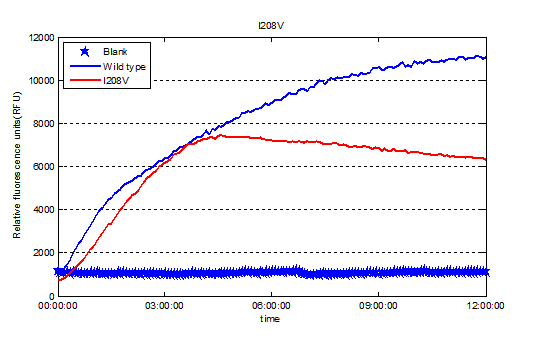
Fig.3.Expression of Plasmid pRset_CFP-1_PETase-I208V.

Fig.3.Relative enzyme activity of Plasmid pRset_CFP-1_PETase-I208V.
Microbial Consortia
xxxxxxxxxx
R-R System
Introduction
R-R (Reporting and Regulation)system is constructed in order to report the expression of PETase gene and regulate the expression process. In this section there are two main concepts we need to prove. The first is whether the ddpX gene can cause cell lysis and the other is whether the ddpX gene can cause cell lysis under the CpxR promoter when inclusion body form led by the overexpression of PETase gene. We constructed the part BBa_K2110008 to prove the validity of this concept.
Constructing Process
This part is modified and improved from the former part BBa_K339007 by changing the original mRFP gene to the novel ddpX gene (Part: BBa_K2110004). The ddpX gene was obtained by colony PCR of E.coli and was verified by sequencing. Considering there is no restriction endonuclease cutting site among the subparts, we used PCR to amplify the CpxR promoter-RBS sequence and linked it with the plasmid backbone and the ddpX gene. Then the new part was obtained.

Proof Result
After we constructed the new biobrick, we firstly used PCR to amplify the whole biobrick and linked it to the recombinant plasmid pUC19 already with PETase gene in it. Then we transfromed the plasmid into E.coli and induced the expression of PETase gene. Then we measured the OD600 of the culture medium. The result is showed below.